 2018-01-21
2018-01-21 2164
2164Пептидная связь
Пептидная (амидная) связь — это вид химической связи, которая возникает вследствие взаимодействия α-аминогруппы одной аминокислоты и α-карбоксигруппы другой аминокислоты. Амидная связь очень прочная, и в нормальных клеточных условиях (37°C, нейтральный ph) самопроизвольно не разрывается. Пептидная связь разрушается при действии на неё специальных протеолитических ферментов (протеаз, пептидгидролаз).
Значение
Пептидные гормоны и нейропептиды, например, регулируют большинство процессов организма человека, в том числе, и принимают участие в процессах регенерации клеток. Пептиды иммунологического действия защищают организм от попавших в него токсинов. Для правильной работы клеток и тканей необходимо адекватное количество пептидов. Однако с возрастом и при патологии возникает дефицит пептидов, который существенно ускоряет износ тканей, что приводит к старению всего организма. Сегодня проблему недостаточности пептидов в организме научились решать. Пептидный пул клетки восполняют синтезированными в лабораторных условиях короткими пептидами.
Белки — органические вещества, в состав молекул которых входят углерод, водород, кислород и азот, а иногда — сера и другие химические элементы.
2. Строение белков. Белки — макромолекулы, состоящие из десятков, сотен аминокислот. Разнообразие аминокислот (около 20 видов), входящих в состав белков.
3. Видовая специфичность белков — различие белков, входящих в состав организмов, относящихся к разным видам, определяемое числом аминокислот, их разнообразием, последовательностью соединения в молекулах белка. Специфичность белков у разных организмов одного вида — причина отторжения органов и тканей (тканевой несовместимости) при их пересадке от одного человека другому.
4. Структура белков — сложная конфигурация молекул белков в пространстве, поддерживаемая разнообразными химическими связями — ионными, водородными, ковалентными. Естественное состояние белка. Денатурация — нарушение структуры молекул белков под влиянием различных факторов — нагревания, облучения, действия химических веществ. Примеры денатурации: изменение свойств белка при варке яиц, переход белка из жидкого состояния в твердое при построении пауком паутины.
5. Роль белков в организме:
— каталитическая. Белки — катализаторы, увеличивающие скорость химических реакций в клетках организма. Ферменты — биологические катализаторы;
— структурная. Белки — элементы плазматической мембраны, а также хрящей, костей, перьев, ногтей, волос, всех тканей и органов;
— энергетическая. Способность молекул белков к окислению с освобождением необходимой для жизнедеятельности организма энергии;
— сократительная. Актин и миозин — белки, входящие в состав мышечных волокон и обеспечивающие их сокращение вследствие способности молекул этих белков к денатурации;
— двигательная. Передвижение ряда одноклеточных организмов, а также сперматозоидов при помощи ресничек и жгутиков, в состав которых входят белки;
— транспортная. Например, гемоглобин — белок, входящий в состав эритроцитов и обеспечивающий перенос кислорода и углекислого газа;
— запасающая. Накопление белков в организме в качестве запасных питательных веществ, например в яйце, молоке, семенах растений;
— защитная. Антитела, фибриноген, тромбин — белки, участвующие в выработке иммунитета и свертывании крови;
— регуляторная. Гормоны — вещества, обеспечивающие наряду с нервной системой гуморальную регуляцию функций организма. Роль гормона инсулина в регуляции содержания сахара в крови.
2 Гетероциклические соединения, органические соединения, молекулы которых содержат циклы, включающие наряду с атомами углерода один или несколько атомов других элементов (гетероатомов). Наибольшее значение имеют гетероциклические соединения, в цикл которых входят атомы N, О, S. К ним относятся многие алкалоиды, витамины, антибиотики, прир. пигменты, они входят в виде структурных фрагментов в молекулы нуклеиновых кислот, белков и других. Гетероциклические соединения - самый многочисленный класс органических соединений, включающий около 2/3 всех известных природных и синтетических органических веществ,
Химические свойства. Для 3- и 4-членных гетероциклических соединений характерна легкость раскрытия напряженного цикла. 5- и 6-членные ненасыщенные гетероциклы (наиболее многочисленный тип гетероциклических соединений), замкнутая сопряженная система связей которых включает (4n + 2) 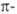 электронов, обладают ароматическим характером (правило Хюккеля) и называют гетероароматическими соединениями. Для них, как и для бензоидных ароматических соединений, Наиболее характерны реакции замещения. При этом гетероатом играет роль "внутренней" функции, определяющей ориентацию, а также активирующее или дезактивирующее влияние на кольцо к действию различных реагентов.
электронов, обладают ароматическим характером (правило Хюккеля) и называют гетероароматическими соединениями. Для них, как и для бензоидных ароматических соединений, Наиболее характерны реакции замещения. При этом гетероатом играет роль "внутренней" функции, определяющей ориентацию, а также активирующее или дезактивирующее влияние на кольцо к действию различных реагентов.
Гетероароматические соединения подразделяют на электроноизбыточные и электронодефицитные. К первым относят 5-членные гетероциклические соединения с одним гетероатомом, в которых секстет 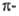 электронов делокализован между пятью атомами цикла, что обусловливает их повышенную активность по отношению к электрофильным агентам. К
электронов делокализован между пятью атомами цикла, что обусловливает их повышенную активность по отношению к электрофильным агентам. К 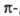 дефицитным относят 6-членные гетероциклы с шестью
дефицитным относят 6-членные гетероциклы с шестью 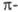 электронами, которые распределяются, как и в случае бензола, между шестью атомами кольца, но один или несколько из них - гетероатомы с большей, чем у углерода, электроотрицательностью. Такие соединения напоминают по реакционной способности производные бензола, несущие ориентанты II рода. Пятичленные гетероароматические соединения с несколькими гетероатомами по формальному признаку можно было бы считать также электроноизбыточными, однако наличие двух и более гетероатомов с их высокой электроотрицательностью, а также способность превращаться в условиях электрофильного замещения в соответствующие катионы обусловливает то, что эти соединения по реакционной способности во многом близки к электронодефицитным гетероциклическим соединениям; их иногда называют
электронами, которые распределяются, как и в случае бензола, между шестью атомами кольца, но один или несколько из них - гетероатомы с большей, чем у углерода, электроотрицательностью. Такие соединения напоминают по реакционной способности производные бензола, несущие ориентанты II рода. Пятичленные гетероароматические соединения с несколькими гетероатомами по формальному признаку можно было бы считать также электроноизбыточными, однако наличие двух и более гетероатомов с их высокой электроотрицательностью, а также способность превращаться в условиях электрофильного замещения в соответствующие катионы обусловливает то, что эти соединения по реакционной способности во многом близки к электронодефицитным гетероциклическим соединениям; их иногда называют  -амфотерными.
-амфотерными.
3 Спирты — органические соединения, содержащие одну или более гидроксильных групп, непосредственно связанных с насыщенным атомом углерода. Спирты можно рассматривать как производные воды, в которых один атом водорода замещен на органическую функциональную группу: R−OH.
Физико-химические свойства спиртов определяются в основном строением углеводородной цепи и функциональной группы −OH, а также их взаимным влиянием:
1) Чем больше заместитель, тем сильнее он влияет на функциональную группу, снижая полярность связи O—Н. Реакции, основанные на разрыве этой связи, протекают более медленно.
2) Гидроксильная группа −ОН уменьшает электронную плотность вдоль прилегающих σ-связей углеродной цепи.
Все химические реакции спиртов можно разделить на три условных группы, связанных с определёнными реакционными центрами и химическими связями:
- Разрыв связи O−H;
- Разрыв или присоединение по связи С−OH;
- Разрыв связи −СOH.
Многие спирты являются незаменимыми участниками биохимических процессов, происходящих в живом организме.
Ряд витаминов можно отнести к классу спиртов:
- Витамин А — ретинол — жирорастворимый витамин, необходимый для нормального обмена веществ.
Витамин B8 — инозит или инозитол — витаминоподобное вещество, участвующее в липидном обмене.
Витамин D — регулирует обмен кальция и фосфора в организме.
Стероидные гормоны, среди которых имеются и спирты, участвуют в регуляции обмена веществ и некоторых физиологических функциях организма.
Полиизопреновый спирт долихол является липидным переносчиком полупродуктов при биосинтезе гликопротеинов.
Среди природных пигментов, участвующих в процессе фотосинтеза и поглощения света, каротиноидов, можно встретить множество соединений с гидроксильными группами.
Примеры фотосинтезирующих каротиноидов, сосредоточенных в хлоропластах растений:
Моносахариды
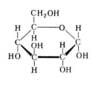
Моносахариды. Циклическая форма
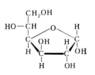
Моносахариды. Циклическая форма
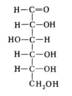
Моносахариды. Ациклическая форма
Моносахариды, органические соединения, одна из основных групп углеводов; содержат гидроксильные группы и альдегидную (альдозы) или кетогруппу (кетозы). М. подразделяют на триозы, тетрозы, пентозы, гексозы и т. д. (3, 4, 5, 6 и т. д. атомов углерода в цепи); природные М. с углеродной цепью, содержащей более 9 атомов углерода, не обнаружены. Для М., содержащих n асимметричных атомов углерода, возможно существование 2n стереоизомеров (см. Изомерия). М. вступают в химические реакции, свойственные карбонильной и гидроксильной группам. Характерная особенность М. — способность существовать в открытой (ациклической) и циклической формах и давать производные каждой из форм; М., содержащие 5-членный цикл, называются фуранозами, 6-членный — пиранозами (см. рис.). М. входят в состав сложных углеводов (гликозиды, олигосахариды, полисахариды) и смешанных углеводсодержащих биополимеров (гликопротеиды, гликолипиды и др.). При этом М. связаны друг с другом и с неуглеводной частью молекулы гликозидными связями. При гидролизе под действием кислот или ферментов эти связи могут рваться с высвобождением М. В природе свободные М., за исключением D-глюкозы и D-фруктозы, встречаются редко. Биосинтез М. из углекислого газа и воды происходит в растениях (см. Фотосинтез); с участием активированных производных М. — нуклеозиддифосфатсахаров — происходит, как правило, биосинтез сложных углеводов. Распад М. в организме (например, спиртовое брожение, гликолиз) сопровождается выделением энергии. Некоторые свободные М. и их производные (например, глюкоза, фруктоза и её дифосфат и др.) используются в пищевой промышленности и медицине.
Конформационное строение
Формулы Фишера и Хеуорса являются условным изображением пространственного строения моносахаридов. Близкое к действительному расположение атомов в пространстве отражают конформационные формулы.
Шестичленный цикл, в котором атомы находятся в состоянии sp3-гибридизации, не может иметь плоскую конформацию, так как это означало бы слишком сильное искажение валентных углов (1200 вместо 1090) и заслоненное положение заместителей. Наиболее выгодной конформацией для большинства шестичленных циклов является конформация “кресла”, в которой все валентные углы равны 1090 и нет заслоненных положений заместителей. Так, у циклогексана есть две энергетически равноценные конформации “кресла”, которые находятся в равновесии. Взаимопревращения между ними называют инверсией цикла.
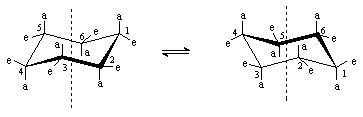
Заместители у каждого атома цикла могут находиться в аксиальном (а) или экваториальном (е) положениях. Аксиальные связи расположены параллельно оси симметрии цикла и направлены попеременно вверх и вниз. Экваториальные связи ориентированы под углом 1090 к оси симметрии цикла также попеременно вверх и вниз. При инверсии цикла экваториальные связи становятся аксиальными и наоборот.
При введении в циклогексан заместителей две конформации кресла становятся энергетически неравноценными. Меньшей энергией, как правило, обладает та конформация, в которой объемистые заместители занимают экваториальное положение. Например, для циклогексанола наиболее выгодной является конформация с экваториальным положением ОН группы:
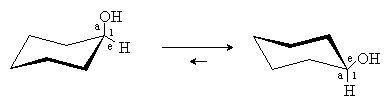
Основой строения пиранозных форм моносахаридов является тетрагидропиран, для которого возможны две энергетически неравноценные конформации кресла.
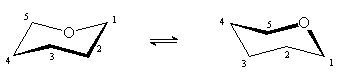
| 1С4 | 4С1 |
Более стабильной является конформация с наименьшим числом объемистых заместителей в аксиальном положении. Для большинства D-альдогексоз это конформация 4С1, в которой группа CH2OH занимает экваториальное положение.
Рассмотрим конформационное строение b-D-глюкопиранозы. Более выгодной для этой формы D-глюкозы является конформация 4С1, в которой все заместители находятся в экваториальном положении.
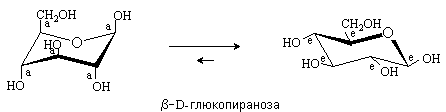
У a-аномера гликозидный гидроксил в этой конформации будет занимать аксиальное положение. Поэтому в равновесной смеси таутомеров D-глюкозы преобладает b-аномер.
b-D-глюкопираноза – единственная D-гексоза с экваториальным положением всех заместителей. Как следствие этого, D-глюкоза - наиболее распространенный в природе моносахарид. Из всего семейства D-альдогексоз в природе встречаются только эпимеры D-глюкозы – D-галактоза и D-манноза, у которых число заместителей, занимающих аксиальное положение минимально.
Моносахариды вступают в химические реакции, свойственные карбонильной и гидроксильной группам. Характерная особенность моносахаридов — способность существовать в открытой (ациклической) и циклической формах и давать производные каждой из форм. Большинство моноз циклизуются в водном растворе с образованием гемиацеталей или гемикеталей (в зависимости от того, являются ли они альдозами или кетозами) между спиртом и карбонильной группой того же самого сахара. Глюкоза, например, легко образует полуацетали, соединяя свои своим С1 и О5, чтобы сформировать 6-членное кольцо, названное пиранозид. Та же самая реакция может иметь место между С1 и О4, чтобы сформировать 5-членное фуранозид.
Химические свойства моносахаридов определяются наличием карбонильной группы (в ациклической форме), полуацетального гидроксила (в циклических формах) и спиртовых ОН групп.
Восстановление
При восстановлении карбонильной группы альдоз образуются многоатомные спирты – глициты.
Окисление
Вследствие своей полифункциональности альдозы окисляются по-разному при действии различных окислителей. При этом может быть окислена карбонильная группа, оба конца углеродной цепи или расщеплена связь С-С.
5 Оксикислоты (оксикарбоновые кислоты, гидрокискарбоновые кислоты) — карбоновые кислоты, в которых одновременно содержатся карбоксильная и гидроксильная группы, например молочная кислота: СН3-СН(ОН)-СООН. Оксикислоты проявляют все свойства, характерные для кислот (диссоциация, образование солей, сложных эфиров и т. д.), и свойства, характерные для спиртов.
Оксикислоты (гидроксикарбоновые кислоты, гидроксикислоты), содержат в молекуле карбоксильную и гидроксильную группы. В статье рассмотрены алифатические оксикислоты. В зависимости от взаимного расположения групп ОН и СООН различают 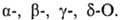 Число групп СООН определяет основность оксикислоты, а число групп ОН (включая ОН в СООН) - их атомность. Многие природные оксикислоты содержат один или несколько асимметрических атомов С и существуют в виде оптических изомеров.
Число групп СООН определяет основность оксикислоты, а число групп ОН (включая ОН в СООН) - их атомность. Многие природные оксикислоты содержат один или несколько асимметрических атомов С и существуют в виде оптических изомеров.
Оксикислоты широко распространены в природе, их остатки входят в состав сфинголипидов животных и растений.
Оксикислоты - кристаллические вещества, низшие оксикислоты из-за сильной гигроскопичности - густые сиропообразные жидкости. хорошо растворимы в воде. Физические свойства некоторых оксикислот представлены в таблице. Оксикислоты вступают в реакции, характерные для карбоновых кислот и спиртов, обладают также специфическими свойствами. Они более сильные кислоты, чем соответствующие карбоновые. Это объясняется существованием внутримолекулярной водородной связи между группами ОН и СООН в 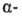 и
и 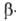 -оксикислоты; более сильную водородную связь образует карбоксилат-анион, получающийся при диссоциации оксикислоты. Повышению кислотности способствует также индуктивный эффект группы ОН.
-оксикислоты; более сильную водородную связь образует карбоксилат-анион, получающийся при диссоциации оксикислоты. Повышению кислотности способствует также индуктивный эффект группы ОН. 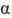 - оксикислоты при нагревании дегидратируются с образованием лактидов.
- оксикислоты при нагревании дегидратируются с образованием лактидов. 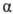 - оксикислоты с третичной группой ОН при нагр. образуют кетоны и
- оксикислоты с третичной группой ОН при нагр. образуют кетоны и  - непредельные кислоты, а при окислении (КМnО4, Н2СrO4 и др.)-кетоны
- непредельные кислоты, а при окислении (КМnО4, Н2СrO4 и др.)-кетоны
Оксокислоты вступают в реакции, характерные для карбоксильной и карбонильной групп. Отличительная черта оксокислот – легкость, с которой протекает их декарбоксилирование.
a -Оксокислоты легко отщепляют СO2 и СО при нагревании в присутствии серной кислоты.

b -Оксокислоты неустойчивы и самопроизвольно декарбоксилируются с образованием кетонов.
CH3COCH2COOH ® CH3COCH3 + CO2
b -Оксокислоты и их эфиры обладают специфическими свойствами, которые связаны с их повышенной СН-кислотностью. Повышенная подвижность протонов метиленовой группы обусловлена электроноакцепторным влиянием двух карбонильных групп. В результате b -оксокислоты существуют в виде двух таутомерных форм: кетонной и енольной (см. лек. №11), причем содержание енольной формы в равновесной смеси значительное. Енольные формы дополнительно стабилизируются за счет наличия в них системы сопряженных p -связей и внутримолекулярной водородной связи.
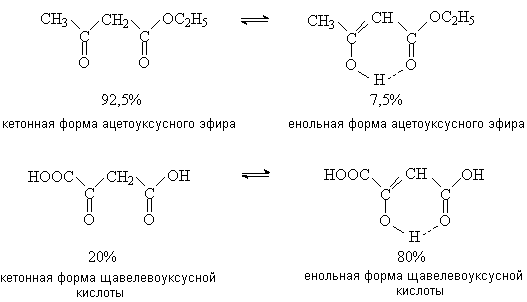
Центральное место среди b -оксокислот и их производных занимает ацетоуксуный эфир (этиловый эфир ацетоуксусной кислоты). Существование в виде двух таутомерных форм обуславливает его двойственную реакционную способность. Как кетон, ацетоуксусный эфир присоединяет нуклеофильные реагенты: HCN, NaHSO3, фенилгидразин. Как енол, присоединяет бром, образует хелатные комплексы с ионами переходных металлов, ацилируется хлорангидридами кислот.
При действии на ацетоуксусный эфир какого-либо реагента в реакцию вступает один из таутомеров. Поскольку второй таутомер за счет смешения равновесия восполняет убыль первого, таутомерная смесь реагирует в данном направлении как единое целое.
Ацетоуксусный эфир широко применяется в органическом синтезе как исходное вещество для получения кетонов, карбоновых кислот, гетерофункциональных соединений, в том числе производных гетероциклов, представляющих интерес в качестве лекарственных средств. Так, производные пиразолона используют как исходные вещества в синтезе ненаркотических анальгетиков – антипирина, амидопирина и анальгина.
Оксикислоты весьма широко распространены в природе. Так, к оксикислотам относятся винная, лимонная, яблочная, молочная и некоторые другие природные кислоты, а их название отражает первичный природный источник, в котором было найдено данное вещество.
6 Ароматические соединения, характеризуются наличием ароматической системы связей (см. Ароматичность). В более узком смысле к ароматическим соединениям относят только бензоидные соединения, т.е. бензол. би-, три- и полициклические соединения, построенные из конденсированных или неконденсированных бензол.ных ядер, и их производные (ароматические углеводороды наз. аренами).
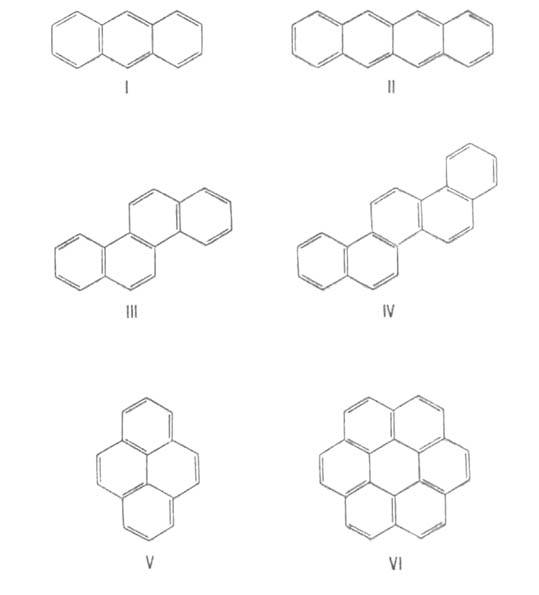
В конденсированных ароматических соединениях два соседних "срощенных" цикла имеют два общих атома. При этом возможны три типа сочленения циклов: линеарное, или линейное, как в антрацене (ф-ла I) и тетрацене (II); ангулярное, или угловое, например в фенантрен., хризене (III), пицене (IV); пери-сочленение, отличающееся наличием атомов С, общих для трех циклов, как в пирене (V) и коронене (VI). В случае пери-сочленения общее число 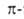 электронов не отвечает правилу Хюккеля (4л + 2) и, следовательно, соответствующие соед. не должны относиться к ароматическим. Однако в них можно выделить отдельные составляющие их моноциклические ароматические соединения (обычно с шестью
электронов не отвечает правилу Хюккеля (4л + 2) и, следовательно, соответствующие соед. не должны относиться к ароматическим. Однако в них можно выделить отдельные составляющие их моноциклические ароматические соединения (обычно с шестью  электронами) или рассматривать
электронами) или рассматривать  электронные оболочки по периметру полициклической системы; если они включают 10, 14, 18 и т.д.
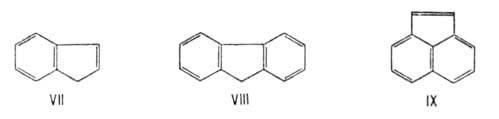
электронные оболочки по периметру полициклической системы; если они включают 10, 14, 18 и т.д.  электронов, то в соответствии с правилом Хюккеля их относят к ароматическим. Таким же образом можно рассматривать конденсированные ароматические соединения, включающие не только шестичленные циклы, например инден (VII), флуорен (VIII), аценафтилен (IX):
электронов, то в соответствии с правилом Хюккеля их относят к ароматическим. Таким же образом можно рассматривать конденсированные ароматические соединения, включающие не только шестичленные циклы, например инден (VII), флуорен (VIII), аценафтилен (IX):
Кроме первых членов ряда, например бензол., нафталина. антрацена, к ароматическим соединениям относятся также их замещенные (гомологи, галогензамешенные, нитросоединения, амины, фенолы. карбонильные соединения и др.). Одним из важных типов ароматические соединения являются жирноароматические соединения - гомологи ароматических соединений и их производные с заместителями в алкильных группах, например бензилхлорид С6Н5СН2Сl, фенилуксусная кислота С6Н5СН2СООН.
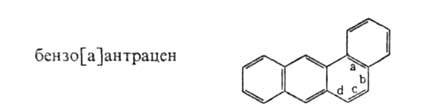
По номенклатуре ИЮПАК и в соответствии с традицией моноциклические ароматические соединения рассматривают как производные бензол.: С6Н5С2Н5-этилбензол, С6Н5Сl - хлорбензол. С6Н5СООН - бензойная кислота, С6Н58О2Сl – бензолcульфохлорид и т.д.; для некоторых сохранены тривиальные назв.: толуол, ксилол, мезитилен, кумол, цимол, стирол, анилин, фенол, крезол. Названия неконденсированных полициклических ароматические соединения строятся по заместительному или соединительному типу, например С6Н5СН2С6Н5 - дифенилметан. С6Н5С6Н5 - бифенил, C6H5C6H4C6H5 - тepенил. Многие конденсир. ароматические соединения имеют тривиальные назв., например нафталин, антрацен, фенантрен. названия более сложных систем основаны на этих тривиальных назв. с добавлением соответствующей приставки и индекса (в квадратных скобках), указывающего место конденсации. например:
Ароматические соединения - жидкости или твердые вещества, отличающиеся от своих алифатических и алициклических аналогов более высокими показателями преломления и поглощением в близкой УФ- или видимой области спектра. Для них характерно наличие т. наз. Магнитного кольцевого тока и поглощение в слабопольной ("ароматической") части спектра ЯМР (область 6,5-8,0 м. д. для 1Н и 110-170 м.д. для 13С).
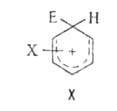
Для ароматических соединений характерны реакции замещения. Наиб. изучено и важно электрофильное замещение, прежде всего галогенирование. нитрование. сульфирование, алкилирование и ацилирование по Фриделю-Крафтсу. Эти реакции облегчаются и направляются преим. в орто- и пара-положения при наличии в ароматические соединения заместителей - ориентантов I рода (Alk, Аг, OR, NR2, SR, F, Cl, Br, I) и затрудняются и направляются преим. в мета-положение ориентантами II рода (COR, COOR, CN, NO2, SO2R, SO3H). Электрофильное замещение осуществляется по механизму "присоединения - отщепления", обычно включающему образование катионного  комплекса (ф-ла X, где X - заместитель в ароматические соединения, ориентант I или II рода; Е – входящая группа), наз. также интермедиатом Уэланда.
комплекса (ф-ла X, где X - заместитель в ароматические соединения, ориентант I или II рода; Е – входящая группа), наз. также интермедиатом Уэланда.
Для ароматические соединения характерно также нуклеоф. замещение при действии N-, О-, S-, С-нуклеофилов, например NR2, RO-, RS-, (RCO)2CH-, а также анионов галогенов (наиб. важны реакции с F ~). При этом замещаемой группой могут служить атомы галогенов, нитро-, амино-, гидрокси-, алкокси-, алкилтио- и сульфогруппы, реже - атомы водорода. Такие реакции часто реализуются в жестких условиях, например щелочное плавление солей сульфокислот проводят при т-pax порядка 300-400°С (в расплаве щелочи при атм. давлении или в водном растворе щелочи при повыш. давлении): ArSO3Na + 2NaOH -> ArONa + Na2SO3 + H2O. Р-ции облегчаются в присутствии соединений Си и особенно при наличии в орто- или пара-положении к уходящей группе ориентантов II рода.
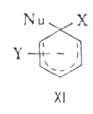
Нуклеофильное замещение может протекать по различным механизмам. Наиболее известный - "присоединение - отщепление", включающий образование анионного 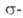 комплекса (XI), наз. комплексом Мейзингеймера. Может осуществляться также радикально-нуклеофильное замещение с промежуточным образованием ароматических анион-радикалов, генерируемых из арилгалогенидов фотохимически или электрохимически:
комплекса (XI), наз. комплексом Мейзингеймера. Может осуществляться также радикально-нуклеофильное замещение с промежуточным образованием ароматических анион-радикалов, генерируемых из арилгалогенидов фотохимически или электрохимически:
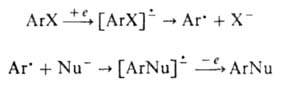
(е- электрон).
Известны также механизмы "отщепления - присоединения". Например, при действии металлич. Li или алкиллития, например BuLi, на о -дигалогенбензолы или при фотолизе о-диазобензойных кислот образуются нестабильные и весьма реакционноспособные дегидроароматич. соед. (арины). Конечный продукт нуклеоф. замещения ArNu образуется в таких случаях в результате присоединения нуклеофила по тройной связи. В случае несимметричных соед. образуются смеси двух продуктов:
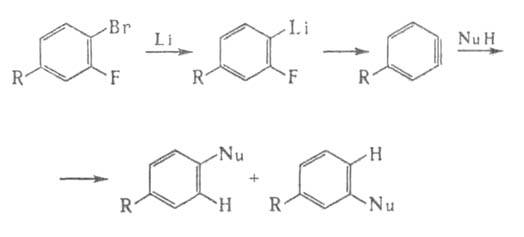 <p.Поляризация тройной связи обеспечивает селективное присоединение нуклеофила к литийаринам, которые образуются, например, при отщеплении сульфинатов Li или производных H2SO3 от 2,6-дилитийсульфонилбензолов: </p.
<p.Поляризация тройной связи обеспечивает селективное присоединение нуклеофила к литийаринам, которые образуются, например, при отщеплении сульфинатов Li или производных H2SO3 от 2,6-дилитийсульфонилбензолов: </p.
Меньшее значение имеет гомолитич. замещение ароматические соединения, например арилирование диазосоединениями и гидроксилирование действием реагента Фентона (Н2О2 + CuSO4 + H2SO4). Ароматические соединения могут подвергаться также прямому металлированию. Особенно важны литирование по механизму протофильного замещения и меркурирование, являющееся реакцией электрофильного замещения. Ароматические соединения могут обменивать галоген на металл (при действии металлов или металлоорганических соед.). С переходными металлами они образуют металлоарены (ареновые 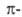 комплексы), напр. дибензолхром.
комплексы), напр. дибензолхром.
Реакции ароматических соединений по замещающим группам в целом подобны реакциям соответствующих алифатических соединений, однако имеются и существенные особенности. Так, ароматические амины образуют с HNO2 устойчивые диазосоединения, способные к азосочетанию и превращающиеся при действии нуклеофилов в разнообразные замещенные ароматические соединения, например по реакции Зандмейера. Двойные соли, образуемые галогенидами арилдиазония и галогенидами различных металлов, при действии порошка Сu, Zn или Bi разлагаются с образованием ароматических металлоорг. соединений (р-ция Несмеянова).
Из реакций присоединения ароматических соединений наиб. важно каталитич. гидрирование - общий метод синтеза соединений ряда циклогексана. Ароматические соединения присоединяют щелочные металлы; образующиеся продукты, например нафтилид натрия 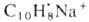 , представляют собой ионные пары катиона металла и анион-радикала ароматических соединений. При действии донора протонов (обычно воды) они превращаются в дигидроароматические соединения. Последние препаративно получают действием на ароматические соединения Li или Na в жидком NH3 в присутствии алканола (р-ция Берна). Менее характерно для ароматические соединения циклоприсоединение. Так, термин. [2+ 4]-циклоприсоединение возможно лишь для активированных ароматических соединений, например 1,2,4,5-тетраметилбензола (дурола) и нафталина, с использованием активных диенофилов; в случае обычных диенофилов требуется УФ-облучение. Последнее приводит к димеризации или изомеризации ароматические соединения, напр.:
, представляют собой ионные пары катиона металла и анион-радикала ароматических соединений. При действии донора протонов (обычно воды) они превращаются в дигидроароматические соединения. Последние препаративно получают действием на ароматические соединения Li или Na в жидком NH3 в присутствии алканола (р-ция Берна). Менее характерно для ароматические соединения циклоприсоединение. Так, термин. [2+ 4]-циклоприсоединение возможно лишь для активированных ароматических соединений, например 1,2,4,5-тетраметилбензола (дурола) и нафталина, с использованием активных диенофилов; в случае обычных диенофилов требуется УФ-облучение. Последнее приводит к димеризации или изомеризации ароматические соединения, напр.:
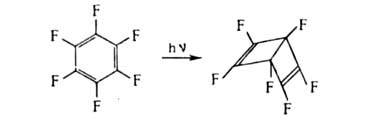
Жирноароматические соединения обычно окисляются по атому С алкильной группы, соседнему с ароматич. кольцом, с сохранением бензольных колец. Таким способом получают ароматические кислоты (напр., терефталевую из n-ксилола), альдегиды (n-нитробензальдегид из n-нитротолуола), кетоны (ацетофенон из этилбензола), спирты (трифенилкарбинол из трифенилметана). Большое практическое значение имеет превращение алкилароматических соединений в гидропероксиды, термическое разложение которых приводит к фенолам и алифатическим карбонильным соединениям, например синтез фенола и ацетона из гидропероксида кумол. (р-ция Сергеева). Конденсированные ароматические системы менее устойчивы к окислению, что используется, например, в синтезе фталевой кислоты из нафталина.
Ароматические углеводороды получают в промышленности из продуктов коксования каменного угля и ароматизацией нефтяных углеводородов, а далее превращаются в разнообразные замещенные. В связи с уменьшением запасов нефти перспективной становится ароматизация алифатических и алициклических углеводородов, получаемых при гидрировании каменного угля и на основе синтез-газа. Лабораторные способы получения ароматические соединения основаны на превращении ароматических углеводородов или др. доступных ароматические соединения; в некоторых случаях используют дегидрирование производных циклогексана, циклотримеризацию ацетиленов и ароматизацию аддуктов, образующихся по реакции Дильса-Альдера. Ароматические соединения - важные промежуточные и целевые продукты промышленного орг. синтеза.
Алкадиены
Строение алкадиенов
Алкадиены — ациклические углеводороды, содержащие в молекуле, помимо одинарных связей, две двойные связи между атомами углерода и соответствующие общей формуле СnН2n-2.
В зависимости от взаимного расположения двойных связей различают три вида диенов:
• алкадиены с кумулированным расположением двойных связей
СН2=С=СН2
• алкадиены с сопряженными двойными связями
CH2=CH—CH=CH2
• алкадиены с изолированными двойными связями
CH2=CH—CH2—CH=CH2
Эти три вида алкадиенов существенно отличаются друг от друга по строению и свойствам. Центральный атом углерода (атом, образующий две двойные связи) в алкадиенах с кумулированными связями находится в состоянии ер-гибридизации. Он образует две Þ-связи, лежащие на одной прямой и направленные в противоположные стороны, и две я-связи, лежащие в перпендикулярных плоскостях. п -Связи образуются за счет негибридизованных р-орбиталей каждого атома углерода.
Свойства алкадиенов с изолированными двойными связями практически ничем не отличаются от свойств алкенов, разве что алкадиены вступают в соответствующие реакции в две ступени. Атомы углерода, образующие двойные связи, находятся в sр2-гибридизации.
Свойства алкадиенов с сопряженными связями весьма специфичны, так как сопряженные л-связи существенно влияют друг на друга.
р-Орбитали, образующие сопряженные п -связи, фактически составляют единую систему (ее называют п -системой), так как р-орбитали соседних л-связей частично перекрываются.
Длины двойных связей (1 и 3) составляют 0,137 нм (двойная связь в алкенах — 0,132 нм), а одинарной (2) — 0,146 нм (0,154 нм у алканов). Таким образом, можно считать, что кратность связей 1 и 3 несколько меньше двух, а связи 2 больше единицы.
Алки́ны (иначе ацетиленовые углеводороды) — углеводороды, содержащие тройную связь между атомамиуглерода, образующие гомологический ряд с общей формулой CnH2n-2. Атомы углерода при тройной связи находятся в состоянии sp-гибридизации.
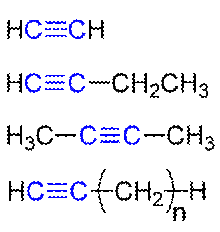

Алкины

3D модель ацетилена — простейшего алкина
Для алкинов характерны реакцииприсоединения. В отличие от алкенов, которым свойственны реакции электрофильного присоединения, алкины могут вступать также и в реакции нуклеофильного присоединения. Это обусловлено значительным s-характером связи и, как следствие, повышенной электроотрицательностью атома углерода. Кроме того, большая подвижность атома водорода при тройной связи обуславливает кислотные свойства алкинов в реакциях замещения.
Алкины по своим физическим свойствам напоминают соответствующие алкены. Низшие (до С4) — газы без цвета и запаха, имеющие более высокие температуры кипения, чем аналоги в алкенах. Алкины плохо растворимы в воде, лучше — в органических растворителях.
8 Жиры и масла, общая характеристика
Жирные масла растений и жиры запасных тканей животных представляют собой наряду с углеводами концентрированный энергетический и строительный резерв жизнедеятельности организма. До 90% видов растений содержат запасные жиры в семенах, но они могут накапливаться и в других органах растений. Основная роль запасных жиров в растении - использование их в качестве резервного материала (во время прорастания семян и развития зародыша); кроме того, они выполняют важную роль защитных веществ, помогающих организмам переносить неблагоприятные условия окружающей среды, в частности низкие температуры. Накапливаясь в семядолях зимующих семян, жиры способствуют сохранению зародыша в условиях мороза. У деревьев умеренного пояса при переходе в состояние покоя запасной крахмал древесины превращается в жир, повышающий морозостойкость ствола.
У животных жиры являются конечными или временными запасными веществами. Конечные запасы, например жир молока, не подлежат использованию самим организмом. Только временные запасные жиры, типичные для жировых тканей, являются мобильными продуктами. Именно эти жиры одновременно являются продуктами, используемыми человеком для пищевых, лекарственных и технических целей.
Строение жиров
Жиры состоят почти исключительно из глицеридов жирных кислот, то есть сложных эфиров глицерина и высокомолекулярных жирных кислот. Глицериды имеют следующую общую формулу:
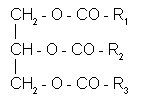
где R1, R2, R3 - радикалы жирных кислот. В природных жирах обнаружено более 200 различных жирных кислот. Преoблaдающими являются жиpныe кислоты с четным числом углеродных атомов от 8 до 24. Жирные кислоты с короткой цепью, содержащей менее 8 углеродных атомов (капроновая, масляная и др.), в составе глицеридов не встречаются, но могут присутствовать в свободном виде влияя на запах и вкус жиров. Большинство жиров содержит 4-7 главных и несколько сопутствующих (составляющих менее 5% от суммы жирных кислот. Достаточно сказать что до 75% жиров составляют глицериды всего трех кислот - пальмитиновой, олеиновой или линолевой.
Входящие в состав триглицеридов жирные кислоты могут быть насыщенными и ненасыщенными. Жиры некоторых растений содержат специфические жирные кислоты, характерные только для этих растений. Так, масло клещевины содержит оксикислоту - рицинолевую; хаульмугровое масло образовано глицеридами циклических кислот - гиднокарповой, хаульмугровой; некоторые кислоты характерны для растений определенных семейств.
Глицериды бывают однокислотные и разнокислотные (смешанные). У однокислотных глицеридов этерификация глицерина произошла с тремя молекулами одной и той же жирной кислоты, например триолеин, тристеарин и т.п. Однако жиры, coстоящие из однокислотных триглицеридов, в природе встречаются довольно редко (оливковое масло, касторовое масло). В образовании жиров доминирует закон максимальной разнородности - подавляющее большинство известных жиров представляют смеси разнокислотных глицеридов (например, стеаринодиолеин, пальмитиноолеинолеин и т.п. В настоящее время известно свыше 1300 различных жиров, различающихся по составу жирных кислот и образуемых ими разнокислотных глицеридов.
Свойства жиров
Свойства жиров определяются качественным составом жирных кислот, их количественным соотношением, процентным содержанием свободных, не связанных с глицерином, жирных кислот, соотношением различных триглицеридов и т.п.
Насыщенные жирные кислоты образуют триглицериды, имеющие при обычной температуре твердую консистенцию. Среди них встречаются как животные (например, говяжий жир), так и растительные (например, масло какао) жиры. Ненасыщенные жирные кислоты образуют триглицериды, имеющие при тех же условиях жидкую консистенцию - животные жиры (например, рыбий жир) и подавляющее большинство растительных масел.
Жиры и масла жирны на ощупь, нанесенные на бумагу, оставляют характерное "жирное" пятно, не исчезающее при нагревании, а, наоборот, еще сильнее расплывающееся. При обыкновенной температуре масла не загораются, но нагретые или в виде паров горят ярким пламенем. Чистые триглицериды бесцветны, но природные жиры более или менее окрашены. Масла обычно желтоватые вследствие присутствия каротиноидов, некоторые из них могут быть окрашены хлорофиллом в зеленый цвет, или, что еще реже, в красно-оранжевый или иной цвет в зависимости от вида липохромов. Запах и вкус свежих жиров специфичны. Запах обусловлен присутствием следов эфирных масел (терпены, алифатические углеводороды и др.). В некоторых жирах содержатся обладающие запахом сложные эфиры низкомолекулярных кислот. Специфический запах рыбьих жиров обусловлен сильно ненасыщенными жирными кислотами или, вернее, продуктами их окисления.
Плотность подавляющего числа жиров находится в пределах 0,910-0,945. Лишь у немногих масел (например, касторового) плотность выше - до 0,970 (при 20°С, по ГФ X).
В воде жиры и масла нерастворимы, но их можно заэмульгировать в воде с помощью поверхностно-активных веществ. В этаноле растворяются трудно (или не растворяются), за исключением касторового масла. Легко растворимы в диэтиловом эфире, хлороформе, сероуглероде, бензине, петролейном эфире, вазелиновом масле. Жиры и масла смешиваются между собой в любых соотношениях. Они являются хорошими растворителями эфирных масел, камфоры, смол, серы, фосфора и ряда других веществ.
Температура плавления твердых жиров возрастает с числом углеродных атомов, входящих в их состав жирных кислот. Поскольку жиры представляют сложные смеси разных триглицеридов, точка плавления их обычно не бывает четко выраженной. Сказанное в равной степени относится и к температуре застывания.
Температура кипения жиров не может быть определена, поскольку при нагревании до 250°С они разрушаются с образованием из глицерина сильно раздражающего слизистые оболочки глаз альдегида акролеина.
Кипят они в высоком вакууме. Жирные масла, состоящие из простых триглицеридов, оптически неактивны, если они не содержат примеси оптически активных веществ. В случае смешанных триглицеридов некоторые жирные масла могут проявлять оптическую активность.
Показатель преломления тем выше, чем больше содержится в жире триглицеридов ненасыщенных кислот. Например, масло какао имеет показетель преломления 1,457, миндальное - 1,470, льняное - 1,482.
Химические свойства жиров проявляются в их способности к омылению, прогорканию, высыханию и гидрогенизации.

