2020-06-30
2020-06-30 236
236Нервно-мышечные заболевания. Классификация. Клиника, диагностика, подходы к лечению.
Классификация
1. Прогрессирующие мышечные дистрофии (первичные миопатии)
2. Спинальные и невральные амиотрофии (вторичные миопатии)
3. Врожденные непрогрессирующие миопатии
4. Нервно-мышечные заболевания с миотоническим синдромом
5. Пароксизмальные миоплегии
6. Миастения
Классификация из лекции:
1. Мышечные дистрофии
2. Врожденные мышечные дистрофии
3. Врожденные (структурные) миопатии
4. Дистальные миопатии
5. Другие миопатии
6. Миотонические синдромы
7. Заболевания мышечных ионных каналов
8. Злокачественная гипертермия
9. Метаболические миопатии
10. Наследственные кардиомиопатии
11. Врожденные миастенические синдромы
12. Болезни моторного нейрона
13. Наследственные атаксии
14. Наследственные моторные и сенсорные нейропатии
15. Наследственные параплегии
16. Другие нервно-мышечные заболевания
Клиника
Жалобы: снижение мышечной силы, изменения походки, ограничение объема движений в конечностях.
Анамнез:
· Период новорожденности: затяжные роды, мышечная гипотония вплоть до симптомокомплекса «вялого ребенка»: необычная поза (поза «лягушки»), снижение сопротивления к пассивным движениям, увеличение объема движений в суставах, снижение общей двигательной активности, задержка моторного развития).
· Раннее развитие: трудности при вскармливании, отставание двигательных навыков (позже удерживает головку, самостоятельно сидит, встает, ходит), трудности при вставании с пола или из положения сидя, подъеме по лестнице, беге.
· У некоторых пациентов может наблюдаться отставание в психическом развитии.
При исследовании родословной выявляются лица с аналогичным заболеванием.
Симптоматика напоминает периферический паралич: мышечные атрофии, гипотония, гипорефлексия. Характерно симметричное поражение мышц и локализация атрофий в определенных группах мышц. Может наблюдаться псевдогипертрофизм. Чувствительность сохранена. Раннее развитие костно-суставных деформаций (контрактуры суставов, сколиоз).
Диагностика
Критерии диагноза:
· Дебют заболевания (когда появились первые симптомы)
· Первоначальная локализация атрофий и дальнейшее распространений
· Темп течения (быстро или медленно прогрессировало заболевание)
· Семейный фон (анализ родословной)
· Дополнительные нарушения (интеллект, костно-суставная, СС системы)
· Данные дополнительных обследований (ДНК-диагностика, ЭМГ, ЭНМГ, ЭКГ, анализы крови)
Методы диагностики
· Биохимический анализ крови (КФК, ЛДГ, АСТ, АЛТ)
· Электронейромиография + электромиография
· Биопсия мышечной ткани (оцениваем морфологию мышечных волокон – атрофия, гипертрофия, повреждение; применяются различные гистохимические окрашивания)
· МРТ мышц
· Генетические методы исследования (полногеномное, полноэкзамное секвенирование, секвенирование по Сангеру, ПЦР, NGS, MLPA)
Подходы к лечению
✓ Коррекция метаболизма скелетной мускулатуры (препараты антагонистов кальция, витамин Е, фосфорсодержащие соединения).
✓ Стимуляция сегментарного аппарата (миостимуляция, нейростимуляция, рефлексотерапия, ЛФК).
✓ Коррекция кровотока (массаж, тепловые процедуры на определенные участки).
✓ Диета (достаточное обеспечение белком, витаминами, минералами).
✓ Ортопедические мероприятия консервативного (специальные шины) и оперативного характера (ахиллотомия, пересечение икроножной мышцы), направленные на коррекцию контрактур и формирующихся патологических установок конечностей. При этом в каждом случае необходимо индивидуально взвесить предполагаемую пользу и возможный вред от оперативного вмешательства.
✓ Симптоматическая терапия при ДН, СН.
Прогрессирующие мышечные дистрофии (ПМД). ПМД Дюшенна и Беккера. Клиника, диагностика, лечение.
Оба заболевания обусловлены мутациями гена дистрофина, расположенного в локусе Хр 21. При дистрофии Дюшенна эта мутация приводит к тяжелому отсутствию (<5%) дистрофина (белок мембраны мышечных клеток). При дистрофии Беккера к образованию аномального дистрофина или его недостаточности.
Х-сцепленное рецессивное наследование. Вместе поражают 5:1000 людей, у большинства ПМД Дюшенна. У женщин-носительниц может быть бессимптомное повышение КФС и, возможно, гипертрофия задней части голени.
ПМД Дюшенна
Болеют мальчики, дебют около 5 лет. Быстрое прогрессирование заболевания – потеря самостоятельной способности двигаться с 8 до 12 лет в среднем. У 30% задержка умственного развития. Прогрессируюшая СН и ДН после 15 лет. Смерть 20-25 лет.
Клиника
· Изменение походки по типу «утиной»
· Затруднения при ходьбе по лестнице, не могут прыгать, приседать, часто падают, получают переломы рук (20%)
· Миопатические приемы (Говерса), вставание «лесенкой»
· Псевдогипертрофия икроножных мышц (замена мышц соединительной и жировой тканью)
· «Крыловидные» лопатки
· Гиперлордоз в поясничном отделе позвоночника, разгибание в коленных суставах, эквино-варусная установка ног
· Контрактуры голеностопных суставов
· Снижение мышечной силы (MRS Scale)
· Снижение тонуса мышц
· Снижение или отсутствие сухожильных рефлексов

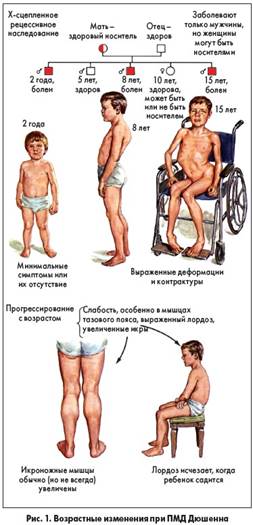
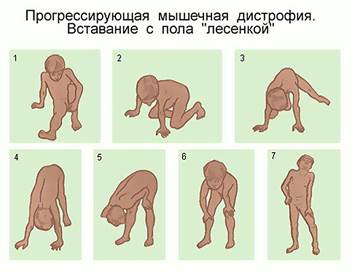
С возрастом сгибательные контрактуры суставов, гиперлордоз, сколиоз прогрессируют. Возникает дыхательная недостаточность, формируется дилатационная кардиомиопатия, затем СН.
Диагностика
Диагноз подозревают, опираясь на клинические признаки, возраст начала, семейный анамнез.
Биохимия крови: повышение КФК (>100 раз), ЛДГ в 3-4 раза, АЛТ, АСТ (десятки раз, имеют внепеченочное происхождение).
УЗИ мышц: признаки мышечной дегенерации – замена мышечной ткани на жировую и фиброзную.
ЭМГ: изменения, характерные для первичного поражения мышц (потенциалы моторных единиц быстро возрастают, имеют небольшую продолжительность и низкую амплитуду).
Молекулярно-генетические методы: обнаружение мутации в Хр 21.
Биопсия: разнокалиберные мышечные волокна, группы некротизированных и регенерирующих волокон, большинство мышечных волокон заменены соединительной и жировой ткани. Подтверждение диагноза – выявление недостаточности дистрофина или его отсутствие (при ПМД Дюшенна чаще отсутствует, Беккера – низкий молекулярный вес или низкая концентрация).
ЭКГ, ЭхоКГ при ПМД Дюшенна: признаки гипертрофической или дилатационной кардиомиопатии.
ПМД Беккера
Болеют мальчики, прогрессирующая мышечная слабость после 6 лет. Прогрессирование заболевания медленнее и легче, чем при ПМД Дюшенна. Способность передвигаться обычно сохраняется до 15 лет, многие могут ходить до взрослого возраста. Большинство доживают до 30-40 лет. Диагностика, клиника, лечение практически такие же, как при ПМД Дюшенна.
Лечение
Диета, обогащенная витаминами, микроэлементами, пищевые добавки, содержащие кальций, витамины группы В, Д.
ГКС терапия продлевает возможность самостоятельно ходить на 2-3 года.
Преднизолон 0,75 мг/кг/сут. Можно Дефлазакорт 0,9 мг/кг/сут. Курсы разные: ежедневно, через день, 10 дней через 10 дней и другие. Показано раннее назначение ингибиторов АПФ и антиаритмической терапии для профилактики дилатационной кардиомиопатии.
Специфического лечения нет. Рекомендовано выполнять легкие активные упражнения во избежание дисфункциональной атрофии или осложнений от гиподинамии. Пассивные упражнения могут продлить период способности к движению. Ортопедические вмешательства должны быть направлены на поддержание функции и предотвращение контрактур.
ДН иногда можно лечить с помощью применения неинвазивной респираторной поддержки (например, назальная маска). Элективная трахеотомия позволяет доживать до возраста старше 20 лет.
Вопрос 128. Прогрессирующие мышечные дистрофии (ПМД). Поясно-конечностные формы ПМД (Эрба-Рота), лице-плече-лопаточная ПМД (Ландузи-Дежерина). Клиника, диагностика, лечение.