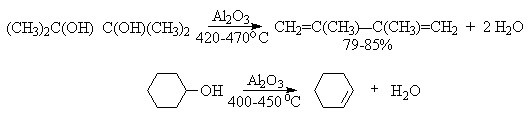2020-09-24
2020-09-24 685
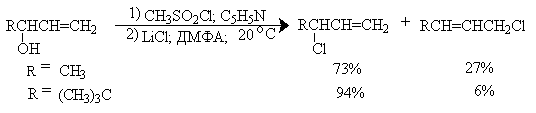
685Тионилхлорид превращает первичные и вторичные спирты в алкилгалогениды с выходом 70-90%. Следует различать две разновидности этой реакции: в присутствии или отсутствии основания (пиридин, триэтиламин, диметиланилин, и др.).

Ранее полагали, что в отсутствии растворителей или в таких растворителях, как петролейный эфир и ароматические углеводороды, замещение гидроксильной группы на хлор с SOCl2 протекает как внутримолекулярное SNi нуклеофильное замещение в хлорсульфите, который, как было установлено, первоначально образуется при взаимодействии SOCl2 со спиртом при низкой (-30оС и ниже) температуре. По этому механизму внутримолекулярное замещение двуокиси серы хлором в хлорсульфите должно происходить с той же стороны, откуда отщепляется уходящая группа SO2. Следовательно, внутримолекулярное нуклеофильное замещение должно сопровождаться полным сохранением конфигурации у асимметрического атома углерода. Для этого процесса был предложен механизм с четырехзвенным переходным состоянием.
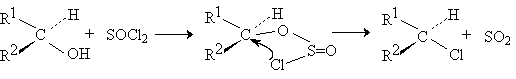
В настоящее время установлено, что процесс такой механизм не реализуется, а реакция протекает с образованием в качестве интермедиата ионных пар.
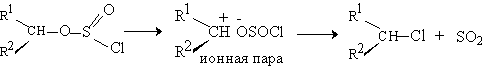
Детальное исследование стереохимии термического разложения оптически активного 2-октилхлорсульфита
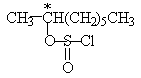
показало явное несоответствие с механизмом SNi. В отсутствие растворителя или петролейном эфире образуется 2-хлороктан с обращением конфигурации, что определенно указывает на замещение хлорсульфитной группы в результате атаки хлорид-ионом с тыла. При термическом разложении 2-октилхлорсульфита в диоксане наблюдается сохранение конфигурации, но этот результат следует рассматривать как следствие двойной инверсии конфигурации. В первой стадии диоксан в качестве нуклеофильного агента замещает хлорсульфит в обычном процессе бимолекулярного нуклеофильного замещения.

Во второй стадии хлорид-ион замещает диоксан в оксониевом катионе.
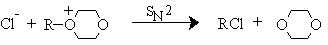
Двойное обращение равносильно сохранению конфигурации у асимметрического атома углерода. Аналогично можно описать превращение спиртов в хлориды и бромиды под действием хлорокиси и бромокиси фосфора.
При замещении гидроксила во вторичных спиртах с помощью тионилхлорида или хлорокиси фосфора в присутствии оснований наблюдается обращение конфигурации. В этом случае имеет место обычное бимолекулярное нуклеофильное замещение в хлорсульфите под действием хлорид-иона как нуклеофила. Источником хлорид-иона служит гидрохлорид третичного амина, образующийся при взаимодействии спирта, тионилхлорида и третичного амина.
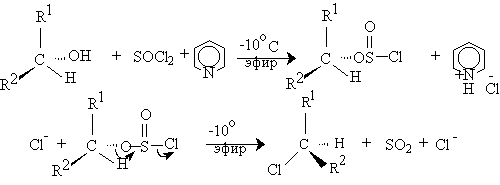
Замещение гидроксила на галоген в реакции спиртов с галогенидами и оксигалогенидами фосфора, мышьяка, серы и селена в присутствии оснований всегда приводит к инверсии конфигурации у хирального атома углерода. Для вторичных спиртов с замещением конкурирует элиминирование с образованием алкенов, которое становится доминирующим для третичных спиртов.
Получение трет-алкилхлоридов и трет-алкилбромидов при взаимодействии третичных спиртов с SOCl2, POCl3, POBr3, PBr5 не эффективно, так как главным направлением реакции становится элиминирование. Трет-алкилгалогениды получаются при взаимодействии спиртов с газообразными HCl и HBr в индифферентном растворителе при 0-10оС.
Перегруппировки и изомеризации при замещении гидроксильной группы первичных и вторичных спиртов на хлор под действием тионилхлорида в присутствии пиридина или другого третичного основания при 0о-(-10о) происходят в значительно меньшей степени по сравнению с замещением с PBr3 или PCl5 в смеси с пиридином.
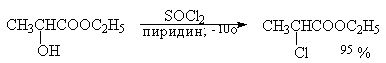
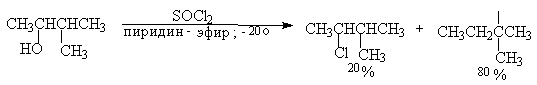
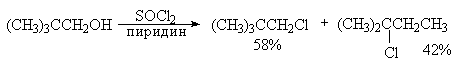
Г. Получение алкилгалогенидов из спиртов и квазифосфониевых солей
Для региоселективного замещения OH-группы в последние годы широкое распространение получили квазифосфониевые соли - аддукты ароматических третичных фосфинов с галогенами, тетрагалогенметанами или N-галогенсукцинимидом. Реакции протекают в мягких условиях и с высокой степенью инверсии конфигурации. Трифенилфосфин образует прочные комплексы с бромом и хлором, которые превращают спирты в алкилгалогениды с высокой региоселективностью. Метод особенно удобен для для вторичных и первичных спиртов, но не эффективен для третичных спиртов, для которых можно ожидать изомеризации и перегруппировки. Иногда в качестве добавки используют пиридин как слабое основание.
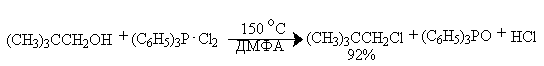
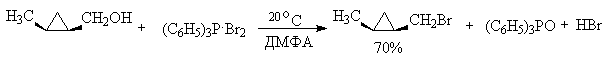
В первой стадии реакции спирт вытесняет галогенид-ион из ковалентно построенного соединения пятивалентного фосфора с образованием ионного фосфониевого интермедиата или ковалентного фосфорана, который затем нуклеофильно атакуется галогенид-ионом с отщеплением трифенилфосфиноксида.
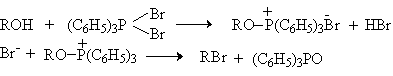
Согласно этой схеме конечным стереохимическим результатом должно быть обращение конфигурации у асимметрического атома углерода, при котором происходит замещение гидроксила на галоген.
Аналогично спирты можно превратить в галогениды с обращением конфигурации при взаимодействии с комплексами трифенилфосфина и тетрагалогенметаном, CCl3CN или CCl3COOEt. В этом случае реакционная способность спиртов уменьшается в ряду:

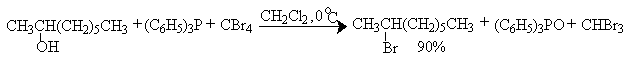
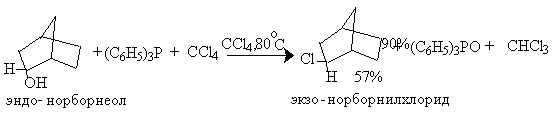
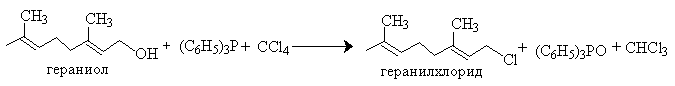
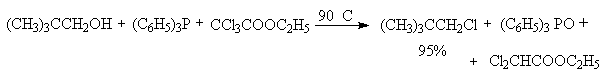

Замещение гидроксила на хлор с помощью систем трифенилфосфин-четыреххлористый углерод и трифенилфосфин - CCl3COOC2H5 в первичных аллиловых спиртах происходит практически без аллильных перегруппировок.
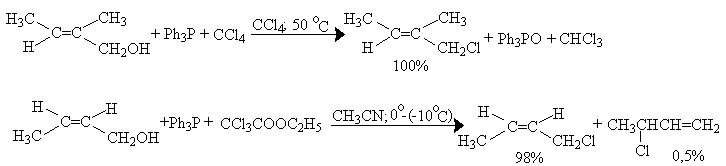
Отсутствие аллильных перегруппировок для первичных аллиловых спиртов выгодно отличает этот способ замещения гидроксильной группы на галоген от других.
Д. Замещение сульфонатной группы в алкилсульфонатах на галоген
Этот метод широко используется для замещения гидроксильной группы в первичных и вторичных спиртах на галогены. Спирты сначала этерифицируют с помощью хлорангидридов сульфокислот, т.к. остаток сульфокислоты является превосходной уходящей группой и легко замещается в мягких условиях под действием галогенид-иона, причем алкилсульфонаты могут быть выделены и очищены. Замещение на галоген обычно осуществляется в ДМФА, ДМСО или ацетоне и сопровождается полной инверсией конфигурации.
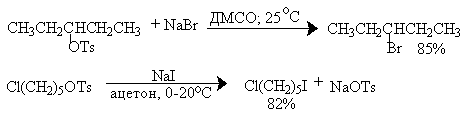
Для вторичных аллиловых спиртов замещение сульфонатной группы на галоген, как правило, сопровождается перегруппировкой, степень которой зависит от структурных факторов и условий проведения реакции (низкая температура способствует большей региоселективности).
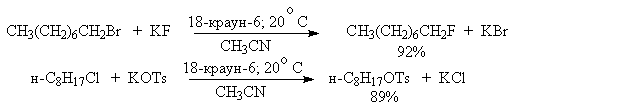
Для получения первичных и вторичных алкилфторидов и, в меньшей степени, алкилхлоридов чрезвычайно эффективным оказался метод межфазного переноса с использованием в качестве катализатора краун-полиэфиров или солей тетраалкиламмония или фосфония. Для получения алкилфторидов в качестве катализатора переноса из твердой фазы в раствор используют краун-полиэфиры. Но для получения хлоридов можно использовать и соли тетраалкиламмония.
Дегидратация спиртов
Протонирование спиртов в ненуклеофильной среде приводит к дегидратации, котоая происходит при нагревании спирта в концентрированной серной, фосфорной кислотах или в суперкислой среде - смеси пятифтористой сурьмы и фторсульфоновой кислоты. Катион алкоксония, отщепляя воду, образует нестабильный интемедиат- карбокатион, который теряет протон с образованием алкена. Наиболее медленная стадия всего процесса - превращение катиона алкоксония в карбокатион. Концентрированная H2SO4 или H3PO4 связывают выделяющуюся воду, что делает весь процесс необратимым.
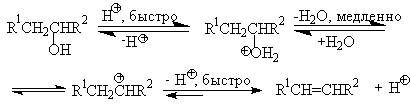
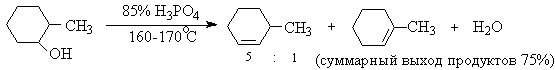
Приведенная последовательность превращений типична для реакций мономолекулярного элиминирования Е1, продукты которого определяются правилом Зайцева, т.е. преобладает наиболее разветвленный при двойной связи алкен. Вторичные спирты подвергаются дегидратациии при нагревании с 85%-ной
Фосфорной кислотой при 160-170оС или с 60-70%-ной серной кислотой при 90-100оС и направление дегидратации соответствует правилу Зайцева.
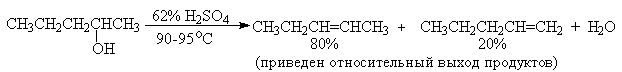
Дегидратацию третичных спиртов можно проводить уже в 20-50%-ной серной кислоте при 85-100oС.
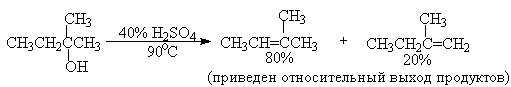
Третичные спирты дегидратируются так легко, что возможна избирательная дегидратация диола, содержащего третичную и первичную гидроксильные группы.
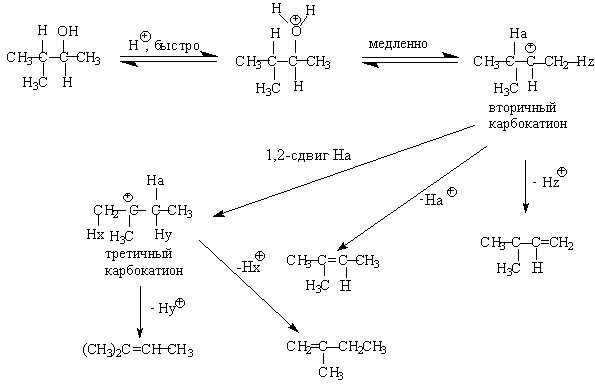
Для El-элиминирования, также как и для других процессов с образованием карбокатиона в качестве интермедиата, характерны перегруппировки, включающие анионотропную 1,2-миграцию гидрид-иона или алкильной группы. В качестве примера можно привести кислотно-катализируемую дегидратацию 3-метилбутанола-2 в 80%-ной серной кислоте.
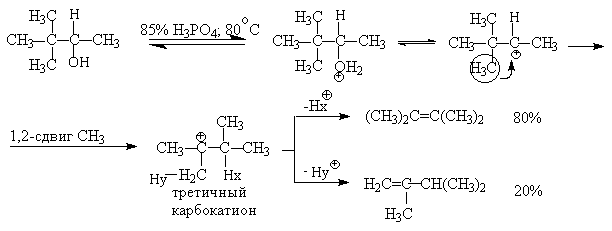
Примером скелетной изомеризации карбокатиона в Е1-элиминировании может служить дегидратация 3,3-диметилбутанола-2.
Для первичных спиртов, вероятно, реализуется иной, Е2 механизм дегидратации в концентрированной серной кислоте, они дегидратируются в гораздо более жестких условиях. Так, пропан ол-1 дает пропилен при нагревании с 96%-ной серной кислотой при 170-190oС, этанол в этих же условиях дает этилен.
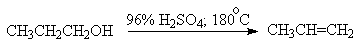
Первичные спирты при взаимодействии с серной кислотой легко образуют полуэфиры серной кислоты. Е2-Элиминированию в этом случае, по-видимому, подвергается полуэфир, а роль основания выполняет гидросульфат-ион или вода.
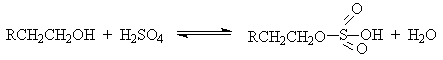
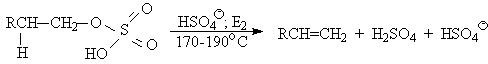
Возможен и другой механизм дегидратации первичных спиртов, в котором субстратом является катион алкоксония, а основанием гидросульфат-ион.
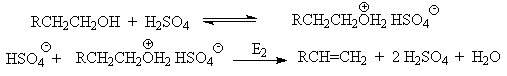
В более мягких условиях при нагревании простейших первичных спиртов с 96%-ной серной кислотой при 130-140oС преимущественно получаются простые эфиры. При этом первичный спирт алкилируется либо под действием полуэфира серной кислоты, либо при взаимодействии с катионом алкоксония.
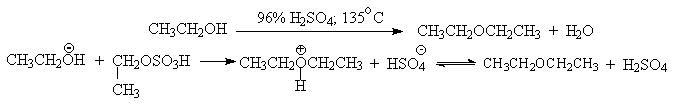
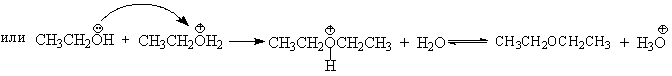
Этим способом получают простейшие простые эфиры - диэтиловый, дипропиловый и дибутиловый эфиры и циклические простые эфиры, например, тетрагидрофуран и диоксан. Вторичные и третичные спирты в этих условиях дегидратируются с образованием алкенов.
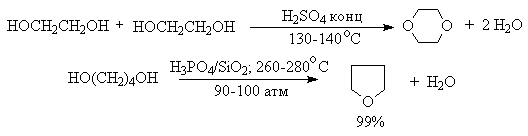
Этот способ неприемлем для получение несимметричных эфиров из двух спиртов, так как при этом образуется смесь трех возможных продуктов ROR, R'OR, R'OR'.
В промышленности для внутри- или межмолекулярной дегидратации вместо серной кислоты в качестве дегидратирующего средства используют безводную окись алюминия. Гетерогенная каталитическая дегидратация первичных, вторичных и третичных спиртов над окисью алюминия при 350-450oС приводит к алкенам.