2020-09-24
2020-09-24 768
768Образование межмолекулярной водородной связи в газовой и конденсированной фазе определяет различие в кислотности первичных, вторичных и третичных спиртов.
В водном растворе и конденсированной фазе кислотность уменьшается в ряду:
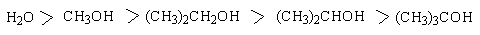
В таблице 1 приведены величины pKa некоторых спиртов, которые наглядно иллюстрируют уменьшение кислотных свойств при переходе от первичных к третичным спиртам.
Существует два различных объяснения влияния заместителей на кислотные свойства спиртов. Одно из них, наиболее традиционное, основано на индуктивном дейстии заместителей. Атом галогена, находящийся при 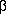 -углеродном атоме спирта, обладает ярко выраженным -I эффектом. Последний поляризацией связей На1-С, С-С, С-0 приводит к поляризации гидроксильной группы и увеличению эффективного положительного заряда на атоме водорода гидроксильной группы. Поляризация гидроксильной группы способствует более легкой диссоциации с образованием алкоголят-аниона и протона. Поэтому р-хлорэтанол должен проявлять свойства более сильной кислоты, чем этанол, а -фторэтанол должен быть более сильной кислотой, чем р-хлорэтанол, так как фтор обладает более сильным -I эффектом, чем хлор. По этой самой причине 2,2,2-трифторэтанол, содержащий три атома фтора, должен быть гораздо более сильной кислотой, чем монофторэтанол. Данные, представленные в таблице 1, согласуются с такой интерпретацией.
-углеродном атоме спирта, обладает ярко выраженным -I эффектом. Последний поляризацией связей На1-С, С-С, С-0 приводит к поляризации гидроксильной группы и увеличению эффективного положительного заряда на атоме водорода гидроксильной группы. Поляризация гидроксильной группы способствует более легкой диссоциации с образованием алкоголят-аниона и протона. Поэтому р-хлорэтанол должен проявлять свойства более сильной кислоты, чем этанол, а -фторэтанол должен быть более сильной кислотой, чем р-хлорэтанол, так как фтор обладает более сильным -I эффектом, чем хлор. По этой самой причине 2,2,2-трифторэтанол, содержащий три атома фтора, должен быть гораздо более сильной кислотой, чем монофторэтанол. Данные, представленные в таблице 1, согласуются с такой интерпретацией.
Таблица 1. Кислотность спиртов в водном растворе
| ROH | pKa | ROH | pKa |
| (CH3)3COH | 18,0 | FCH2CH2OH | 13,9 |
| (CH3)2CHOH | 17,1 | CF3CH2OH | 12,4 |
| CH3CH2OH | 15,9 | CF3CH2CH2OH | 14,6 |
| CH3OH | 15,5 | CF3CH2CH2CH2OH | 15,4 |
| HOH | 15,7 | (CF3)3COH | 5,4 |
| ClCH2CH2OH | 14,3 |
Индуктивный эффект имеет тенденцию к затуханию, если атом галогена более удален от гидроксильной группы. Действительно, 3,3,3-трифторпропанол оказывается более слабой кислотой, чем трифторэтанол, а 4,4,4-трифторбутанол практически не отличается по кислотности от незамещенного первичного спирта. Другое объяснение изменения кислотности спиртов никак не связано с индуктивным влиянием заместителей. Это объяснение основывается на стабильности алкоголят-анионов, образующихся при диссоциации спиртов. Анионы 2-фторэтанола и, особенно, 2,2,2-трифторэтанола гораздо более стабильны, чем незамещенный этилат-ион, так как положительный конец диполя связи C+d-F-d расположен ближе к отрицательно заряженному атому кислорода, чем его отрицательный конец. Поэтому электростатические силы притяжения преобладают над отталкиванием двух одноименно заряженных частиц, а это стабилизирует анион F3ССН3СO- по сравнению с анионом СН3СН2СO-
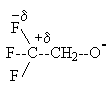
Приведенное объяснение носит название "эффекта поля" и оно не требует привлечения особых, часто умозрительных химических понятий типа нуклеофильного, мезомерного и других эффектов. Алкильные группы, обладающие +I эффектом, дестабилизируют алкоксид-ион и рКа третичного бутилового спирта в воде выше, чем для первичных и вторичных спиртов.
Спирты как слабые ОН-кислоты реагируют со щелочными, щелочноземельными металлами, алюминием, галлием, таллием с образованием ионных или ковалентных алкоголятов.
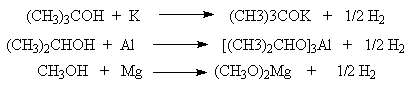
Алкоголяты могут быть получены при действии на спирты сильных оснований - гидридов или амидов щелочных или щелочноземельных металлов, а также реактивов Гриньяра.
Спирты проявляют свойства не только слабых кислот, но и слабых оснований Льюиса, т.е. они обладают амфипротонными свойствами. Как основания Льюиса спирты образуют донорно-акцепторные комплексы с галогенидами и оксигалогенидами фосфора, серы или с другими кислотами Льюиса. С сильными минеральными кислотами спирты образуют соли алкоксония. На этих свойствах основаны многие важные реакции замещения гидроксильной группы на галоген, дегидратации и этерификации спиртов с помощью органических и минеральных кислот и их производных.
2. Замещение гидроксильной группы на галоген
Замещение гидроксильной группы на галоген относится к числу наиболее важных реакций органического синтеза. Существует большое число методов замены гидроксильной группы на галоген. Они различаются регио- и стереоселективностью, а выходы спиртов коляблются в весьма широких пределах.
А. Получение алкилгалогенидов из спирта и галогеноводородов
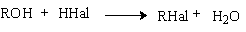
Реакционная способность галогеноводородов уменьшается в ряду
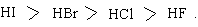
Скорость реакции с HF слишком мала для прямого превращения спиртов в алкилфториды. Скорость реакции замещения резко снижается в ряду
 .
.
Для получения третичных алкилгалогенидов обычно достаточно насытить третичный спирт газообразным галогеноводородом при 0-10о или обработать водной соляной или бромистоводородной кислотами в течение короткого времени при 0-20о. Для получения первичных и вторичных алкилбромидов и алкилиодидов обычно требуется нагревание смеси спирта, концентрированной бромистоводородной кислоты и концентрированной серной кислоты в течение нескольких часов. Вместо концентрированных водных растворов HBr можно использовать бромиды натрия и калия и концентрированую серную кислоту.
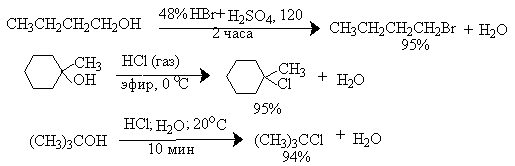
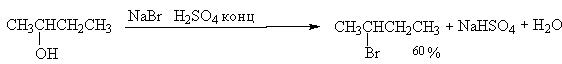
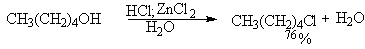
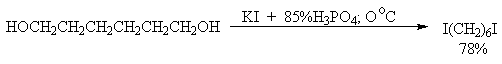
Спирты как слабые основания Льюиса при протонировании образуют соли алкоксония. Протонирование гидроксильной группы превращает плохую уходящую группу OH в хорошую уходящую группу - воду.
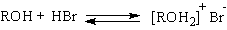
Механизм последующего нуклеофильного замещения зависит от природы радикала спирта. Для первичных спиртов реализуется SN2 - механизм замещения воды в катионе алкоксония на галоген.
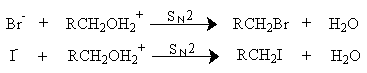
Хлорид-ион в гидроксилсодержащих растворителях сильно сольватирован и проявляет свойства более слабого нуклеофила по сравнению с бромид- и иодид-ионами. Поэтому для получения алкилхлоридов при взаимодействии первичных спиртов с соляной кислотой используют электрофильный катализатор - безводный хлорид цинка. Смесь соляной кислоты и хлорида цинка носит название реактива Лукаса. Хлорид цинка как жесткая кислота Льюиса координируется по атому кислорода, облегчая тем самым замещение гидроксильной группы.
Реакционная способность спиртов по отношению к реактиву Лукаса уменьшается в ряду
Третичные и, по-видимому, вторичные спирты взаимодействуют с галогеноводородами по механизму SN1 с образованием карбокатиона в качестве интермедиата. При этом в реакционной смеси в высокой концентрации находится галогенид-ион - более сильный нуклеофильный агент, чем вода. Поэтому карбокатион стабилизируется не выбросом протона или рекомбинацией с молекулой воды, а взаимодействием с галогенид-ионом как наиболее сильным нуклеофилом.

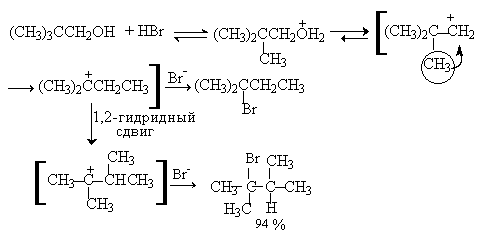
Характерной особенностью процессов с участием карбокатионов являются перегруппировки. Эта реакция может также сопровождаться перегруппировками. Атом водорода при соседнем атоме углерода может мигрировать в виде гидрид-иона к карбокатионному центру. Это смещение называется 1,2-гидридным сдвигом. Роль подобным перегруппировок возрастает тогда, когда в результате гидридного сдвига образуется более стабильный карбокатион.
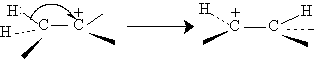
Так, например, при нагревании 3-метилбутанола-2, насыщенного газообразным бромистым водородом, в качестве единственного продукта реакции образуется 2-бром-2-метилбутан вместо нормального продукта замещения гидроксила на бром - 2-бром-3-метилбутан. Ниже приведена предполагаемая последовательность превращений, включающая изомеризацию вторичного крбокатиона в более стабильный третичесый карбокатион за счет 1,2-гидридного сдвига.
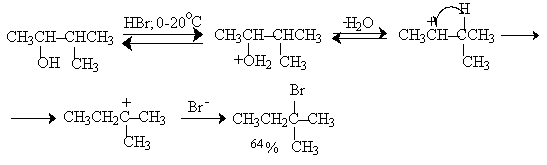
Перегруппировка может наблюдаться и в том случае, если два карбокатиона близки по стабильности. При нагревании с бромистоводородной кислотой пентанол-2 и пентанол-3 образуют смесь 2-бром- и 3-бромпентана за счет перегруппировки двух вторичных карбокатионов.
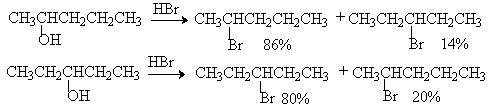

Перегруппировка может происходить не только за счет 1,2-миграции гидрид-иона, но также в результате скелетной изомеризации, когда мигрирует алкильная группа. Так, например, если структурные факторы способствуют образованию третичного карбокатиона, первичный спирт претерпевает перегруппировку Вагнера-Меервейна.
Пентанол-1 в тех же условиях образует только 1-бромпентан без продуктов перегруппировки и это превращение протекает по SN2-механизму. Однако первичные спирты с разветвленной цепью дают некоторое количество изомерного галогенида.
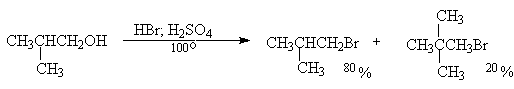
Таким образом, замещение гидроксильной группы спиртов на галоген под действием галогеноводородов без изомеризации осуществляется только для третичных спиртов и нормальных алканолов-1. Для получения индивидуальных алкилгалогенидов из спиртов, способных к изомеризации, следует применять другие методы.
Б. Получение алкилгалогенидов из спиртов и галогенидов фосфора
Для превращения спиртов в алкилгалогениды применяют различные тригалогениды, пентагалогениды и оксигалогениды фосфора: PBr3, PCl5, PCl3, POCl3, PI3 (получается из красного фосфора и иода непосредственно во время реакции). Для получении первичных и вторичных галогенидов на три моля спирта можно расходовать только один моль трехбромистого или трехиодистого фосфора.
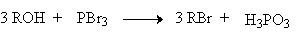
Механизм замещения гидроксильной группы первичных или вторичных спиртов на бром или иод в реакциях с PBr3 или PI3 в точности не установлен. Наиболее разумное предположение заключается в том, что первоначально из спирта образуется трииалкилфосфит (RO)3P и три молекулы HX. Далее происходит постадийное нуклеофильное замещение фосфитной группировки в протонированной форме фосфита под действием галогенид-иона.
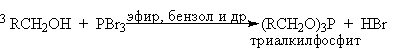
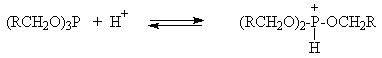
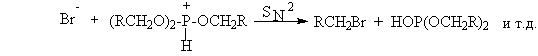
Замещение двух других фосфитных группировок происходит по аналогичному механизму. Реакция катализируется третичными аминами: пиридином, N,N-диметиланилином, триэтиламином и др. и для вторичных спиртов сопровождается 100%-ной инверсией конфигурации асимметрического атома углерода. Принимая во внимание, что PI3 не стабилен даже при 20оС, замещение гидроксильной группы на иод проводят при взаимодействии первичного или вторичного спирта, красного фосфора и иода.
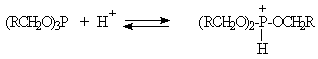
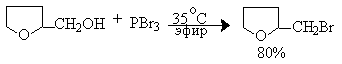
При обработке первичных спиртов PCl3 образуются соответствующие диалкилфосфиты и лишь одна молекула спирта превращается в алкилхлорид; вторичные спирты при этом, в основном, дегидратируются.
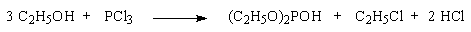
Предполагают, что сначала образуется триалкилфосфит, который только частично расщепляется выделяющимся хлористым водородом.
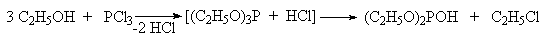
Если проводить эту реакцию в присутствии оснований, связывающих выделяющийся хлористый водород, то образуются только триалкилфосфиты.

Вторичные спирты в реакции с PCl3 подвергаются, главным образом, дегидратации с образованием алкенов. Это неудивительно, если принять во внимание, что галогениды и оксигалогениды фосфора проявляют свойства типичных кислот Льюиса, способствующих этому нежелеательному процессу.
Замещение гидроксильной группы под действием РВгз и других галогенидов и оксигалогенидов фосфора происходит с инверсией конфигурации у асимметрического атома углерода, связанного с гидроксильной группой и часто сопровождается перегруппировками. Так, пентанол-3 в реакции с трехбромистьш фосфором в эфире образует 85% 3-бромпентана и 15% 2-бромпентана - продукта перегруппировки. З-Метилбутанол-2 и неопентиловый спирт с трехбромистьм фосфором также дают смесь двух изомерных галогенидов, в которых доминирует продукт изомеризации.
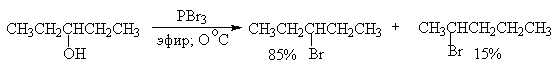
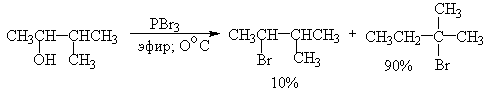
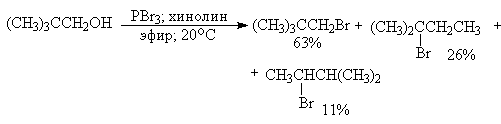
Применение третичных оснований, таких, как пиридин, хинолин, N,N-диметиланилин в смеси с РВг3, РI3, РС15, РОС15, несколько уменьшает долю продукта перегруппировки, но не устраняет ее полностью. Таким образом, галогениды и оксигалогениды фосфора не могут быть рекомендованы в качестве региоселективных или региоспецифических реагентов для замещения гидроксильной группы в спиртах.