2020-10-11
2020-10-11 254
254Физиологическую ак-ть хим-х соед-й открывали случайно или путём перебора: химики-органики синтезировали разнообразные типы орг-х соед-й и передавали их биологам на тестирование. Хотя подобный подход вряд ли можно назвать научным, тем не менее с его помощью находили и находят исключительно акт-е структуры и удачные лекарства. Если посчитать, сколько всего структур может сущ-ть в орг-й химии (перебор комбинаций атомов кислорода, углерода, водорода и азота), то получается около 10180 вещ-в. Теоретически каждое из них можно синтезировать и испытывать, если для этого хватит атомов во Вселенной.
Дело в том, что химики, биологи и медики говорят совершенно на разных языках. Например, медик просит сделать препарат для понижения кровяного давления, а биохимик предлагает найти ингибитор ангиотензинконвертирующего фермента, ликвидация ак-ти к-го и приведет к снижению давления.
Язык химиков – структурные формулы, поэтому для них такая постановка задачи неприемлема. В ответ они не могут предложить ни конкретную структуру, ни даже класс требуемого соед-я. Чтобы создание такого лекарства стала возможным, нужен переводчик биохим-й (или фармакологической) инфо на язык структурных формул. Роль такого переводчика как раз и играет сформировавшаяся в последние десятилетия поддисциплина орг-й химии, получившая название медицинская химия.
Основная задача мед-й химии: создание соед-й с заранее заданной физиологической ак-ю, так называемый рациональный драг-дизайн.
Стратегию рационального дизайна лекарств можно условно разбить на 3 стадии:
1. Поиск или конструирование соединений-лидеров
2. Оптимизация соединения-лидера
3. Разработка лекарства
Соединение-лидер – это хим-е соед-е, к-е имеет желаемую, интересную, но не оптимальную ак-ть. Это структурный прототип будущего лекарства. На первом этапе задача создания лекарства как раз и сводится к тому, чтобы найти его прототип (если, конечно, он не был найден случайным образом).
N-сукцинилпролин – соединение-лидер при создании препарата каптоприла, понижающего кровяное давление.
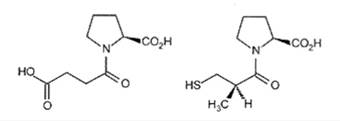
Чтобы искать соединение-лидер, нужно знать его биомишень, то есть макромолекулу в орг-ме человека, на к-ю наше будущее лекарство должно воздействовать, связываясь с ней. В подавляющем большинстве случаев такой мишенью бывает белок (обычно рецептор или фермент), но это может быть и молекула ДНК, и др биомолекулы. Стратегия поиска лидеров зависит от того, что известно о его биомишени, а также от того, что известно о структурах уже существующих лигандов, к-е с ней связываются. Здесь возможно несколько вариантов. Если исследователю не известно ничего – ни структура биомишени, ни структура лиганда, то для поиска соединения-лидера используют метод комбинаторной химии (синтез библиотек соед-й и их тестирование). Фактически это то же самое, что делали раньше, только на новом технологическом, машинном уровне. Химики синтезируют параллельно несколько вещ-в и быстро тестируют их на биомишени с применением современной робототехники.
Когда структура мишени известна, а структуру её лиганда мы не знаем, учёные используют методику, к-я называется de novo дизайн. Создают компьютерную пространственную модель молекулы-мишени, в том числе той её полости, с к-й должно связываться лекарство. Потом на компьютере же совмещают эту полость с различными молекулами – кадидатами на роль лидера(эта процедура называется докинг, по аналогии с заходом корабля в док). При этом структуру гипотетических лидеров нужно подбирать таким образом, чтобы, во-первых, добиться хорошего совмещения размеров молекулы с размером полости, во-вторых, увеличить взаимное связывание молекулы в полости мишени (за счёт слабых взаимодействий: водородных связей, электростатического притяжения, липофильных взаимодействий и т.д.). В результате можно подобрать структуру определённого размера и геометрии, к-я хорошо подходит под мишень. Смоделированное соед-е синтезируют, испытывают на ак-ть и, если атковвая обнаружится, берут его в качестве соединения-лидера.
Когда соединение-лидер найдено, начинается второй этап конструирования лекарства – оптимизация. Нужно так изменить соединение-лидер, чтобы оно имело нужную ак-ть, селективность, растворялось в том, в чём удобно, не было токсичным. Естественно, для этого надо менять его структуру. На практике химики синтезируют структурные аналоги соединения-лидера и тестируют их. Основная проблема на этой стадии заключается в том, что теоретически количество возможных аналогов огромно. Это значит, что и здесь необходимо применять рациональный подход, позволяющий предсказывать, какие именно аналоги нужно синтезировать. Для этого можно использовать опять же компьютерное моделирование, то есть докинг небольшого количества аналогов соединения-лидера с известной ак-ю. С его помощью удается понять, как расположены друг относительно друга хим-е группы, важные для связывания с мишенью, а значит, сократить количество синтезируемых аналогов.
В том случае, когда докинг невозможен, потому что неизвестна структура мишени, а есть только инфо, что у каких-то вещ-в есть нужная ак-ть, обычно используют метом QSAR. Это направление возникло на стыке орг-й химии, матем-го моделирования и компьютерной химии. Дословный перевод: количественное соотношение структура-св-во.
Завершающая стадия создания лек-го соед-я – его разработка. Оптимизированный лидер ещё улучшает таким образом, чтобы он стал удобным для клинического использования и приобрел нужные фармакокинетические характеристики. Часто на этой стадии структуру активных соединений снова изменяют. Здесь много методов с красивыми названиями: создание биоизостеров, пролекарств, пептидомиметиков и т.д. Это сугубо «медхимические» понятия.
Пролекарства – это вещества, не обладающие выраженной физиологической активностью, но способные превратиться в лекарства уже в организме человека. Происходит это в результате либо ферментативной реакции, либо химической (без участия белкового катализатора). Чтобы получить пролекарство, обычно модифицируют какую-то реакционноспособную группу в физиологически активном соединении так, чтобы эта связь разрушилась в организме. С помощью лекарств можно, например, продлить действие препарата, повысить его растворимость в воде и даже изменить его вкус.
Важный метод этого этапа – изостерическая и биоизостерическая замена. Молекулы или ионы, к-е содержат одинаковое число атомов и имеют одинаковое кол-во и расположение электронов. Соответственно изостерическая замена в конструируемом лекарстве - это замена атома или группы на похожую по размеру или валентности. Если при этом сохраняется физиологическая биоизостерической. Интересно, что термин биоизостер относится к соед-ям, получающимся путём замены на совершенно непохожие группировки,но с сох-ем био-х св-в. С помощью биоизостерической замены исследователям удаётся, например, уменьшить токсичного активного соед-я, повысить его усстойчивость к действию ферментативных систем орг-ма и т.д.
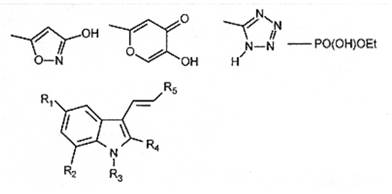
· Примеры группировок, к-е кажутся непохожими на карбоксильную группу (-СООН), но тем не менее часто исп-ся вместо неё при биоизостерической замене.
Естественно, каждая структурная модификация, направленная на улучшение фармакокинетических св-в вещ-ва, приводит к созданию нового хим-го соед-я. А оно, конечно же, может иметь меньшую ак-ть или вообще другой тип ак-ти. Поэтому исследования, посвященные разработке лекарства, часто неотделимы от стадии оптимизации с исп-ем QSAR и компьютерного моделирования.