 2014-02-02
2014-02-02 3622
3622Г
Остатки лизина и 5-гидроксилизина подвергаются окислению постепенно, поэтому количество поперечных связей между отдельными молекулами тропоколлагена, соседними микрофибриллами и пучками волокон с течением времени увеличивается, прочность коллагена возрастает: идёт процесс «созревания».
Учитывая роль ЛИЗ в организации различных вариантов ковалентных сшивок в коллагеновой матрице костной ткани, зубного дентина и цемента становится понятной материальная основа применения лизина как средства, предотвращающего заболевания зубов.
Коллаген активно обновляется, имеет высокие уровни биосинтеза и катаболизма.
Коллагеназа (относится к ММП – см в конце лекции) выделена из кости, десны, периодонта, хряща, кожи, роговицы глаза, синовиальной оболочки, эпителиальных клеток печени, нейтрофилов, эозинофилов, макрофагов, фибробластов, тромбоцитов и т.д. Для действия коллагеназы необходимы ионы Са2+, рН=7-9. Коллагеназа действует специфично: перерезает все три пептидные цепи коллагена в одном месте (≈ на 1/4 расстояния от С-конца между остатками глицина и лейцина) и облегчает дальнейшее действие других протеаз. При разрушении коллагена освобождается гидроксипролин, часть которого выводится с мочой. Количество этой аминокислоты и активность коллагеназы отражают скорость катаболизма коллагена (маркёры распада коллагена).
Коллагеновые болезни (нарушен обмен коллагена)
1) Несовершенный остеогенез – мутации (более 160) в гене, кодирующем синтез коллагена I. Самая неблагоприятная замена глицина на другую аминокислоту – не образуется нормальная тройная спираль. Признаки: ломкость костей, аномалии зубов, треугольная форма лица, гиперподвижность суставов, голубые склеры.
2) Болезнь Книста – дефект коллагена II –укорочение цепей. Укорочение и деформация конечностей, тугоподвижность суставов, кифосколиоз, миопия высокой степени.
3) Синдром Вагнера – дефект коллагена II – в стекловидном теле синтезируется половина молекулы коллагена – прогрессирующая миопия, отслойка сетчатки, патология суставов
4) Синдром Элерса-Данлоса – дефект коллагена III – При врожденном дефиците проколлагеновой пептидазы нарушается формирование всего коллагенового волокна с соответствующими нарушениями формирования скелета, зубного ряда и т.д. Не отщепляются концевые пептиды, нарушается фибриллобразование, поэтому коллаген представлен в виде сети. У этих больных коллаген отличается необычайно высокой растворимостью, в результате изменений в соединительной ткани больные обладают хрупкой кожей и чрезмерно подвижными суставами + спонтанные разрывы крупных сосудов, перфорация кишечника, разрывы беременной матки, спонтанный пневмоторакс.
5) Синдром Альпорта – дефект коллагена IV - нарушение образования базальных мембран. Поражения почек, гематурия и протеинурия.
6) Синдром Гудпасчера - дефект коллагена IV- образование антител к коллагену. Гломерулонефрит, легочный гемосидероз.
7) Буллезный эпидермолиз - дефект коллагена VII - снижение количества заякоренных фибрилл. Эпидермис слабо связан с дермой, легко слущивается и образует пузыри (буллы), которые легко травмируются, эрозируются.
Другие врожденные заболевания:
8) Синдром Морфана – повышенная растворимость коллагеновых волокон, они легко катаболизируются. В моче повышается концентрация пролина.
9) Синдром чрезмерных кожных складок.
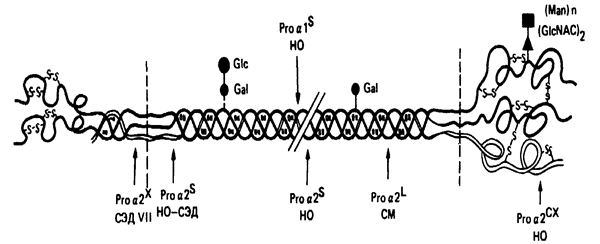
Сайты мутаций в молекуле проколлагена типа I. СЭД (синдром Элерса-Данлоса), СМ (синдром Марфана), НО (несовершенный остеогенез)

