 2014-02-12
2014-02-12 1234
1234Фаренгейта, Цельсия и Кельвина
| Наименование | Шкалы | ||
| параметра | Фаренгейта | Цельсия | Кельвина |
| Абсолютный нуль | -460оF | -273оС | 0 К |
| Точка замерзания воды | 32 оF | 0 оС | 273 К |
| Средняя комнатная температура | 68 оF | 20 оС | 293 К |
| Нормальная температура тела человека | 98,6 оF | 37 оС | 310 К |
| Точка кипения воды | 212 оF | 100 оС | 373 К |
Таблица 2.3
| Равновесное состояние | Температура | |
| Т68, К | t68, оС | |
| Тройная точка равновесного водорода | 13,81 | -259,34 |
| Точка кипения водорода при давлени 33330,6 Па (25/76 нормальной атмосферы) | 17,042 | -256,108 |
| Точка кипения равновесного водорода при 1 атм | 20,28 | -252,87 |
| Точка кипения неона при 1 атм | 27,402 | -246,048 |
| Тройная точка кислорода | 54,361 | -218,789 |
| Точка кипения кислорода при 1 атм | 90,138 | -182,962 |
| Тройная точка воды | 273,16 | 0,01 |
| Точка кипения воды при 1 атм | 373,15 | 100,0 |
| Точка затвердевания цинка | 692,73 | 419,58 |
| Точка затвердевания серебра | 1235,08 | 961,93 |
| Точка затвердевания золота | 1337,58 | 1064,43 |
Как показано на рис.2.1, можно говорить о величине абсолютного нуля температур. При этом за величину абсолютного нуля температур в системе СИ следует принимать температуру 0К, или -273,16 0С.
Как будет показано ниже, по результатам исследования идеального цикла Карно, можно придти к обобщенному термодинамическому понятию температуры. Этот анализ подтвердит соответствие шкал температур, полученных на основе различных свойств веществ, с абсолютной термодинамической шкалой.
Температура является одним из важнейших параметров состояния термодинамической системы.
2.2. Теплоемкость веществ.
Важной характеристикой веществ является теплоемкость. Под теплоемкостью понимают количество тепла, которое потребно для нагревания 1 кг вещества на один градус. Представляется безусловным, что чем больше масса вещества, тем больше энергии требуется для нагревания. Ученые установили, что количественно процесс нагревания тела описывается зависимостью:
ΔQ=m·c·Δt (2.9)
Здесь
ΔQ – количество тепла, кал(ккал) или Дж
m – масса вещества, кг
c – теплоемкость вещества, ккал/кг·град или Дж/кг·град
Δt – изменение температуры тела, град
Отсюда теплоемкость
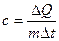 (2.10)
(2.10)
Ученые установили, что количество тепла при изменении температуры на один градус зависит от условий подвода – при постоянном объеме или при постоянном давлении. Соответственно различают две теплоемкости вещества:
- при постоянном объеме - сV
- при постоянном давлении - ср.
Их природа определяется строением вещества и молекулярным весом.
Ученые выяснили, что для одного моля вещества имеется строгая связь (уравнение Майера):
ср - сV = R, (2.11) где R – газовая постоянная этого вещества.
Важную роль, как это будет показано ниже, играет отношение этих двух теплоемкостей: k= 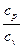 (2.12)
(2.12)
Значение отношения теплоемкостей играет важную роль при изучении адиабатных (т.е. происходящих без обмена энергией с окружающей средой) процессов расширения и сжатия газов. Величину k называют показателем адиабаты. Уравнение адиабатного процесса записывают в виде
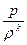 = const
= const
Молекулярно-кинетическая теория газов позволяет создать простую модель определения теплоемкости. Ученые предположили, что при нагревании газов происходит увеличение энергии нескольких видов движения молекул. Рассмотрим суть рассуждений на примере одномолекулярного газа, например, простейшего из них – гелия. Положив, что молекулы гелия – это упругие шарики, имеющие возможность перемещаться в направлении трех осей координат, следует считать, что кинетическая энергия при изменении температур должна делиться на три доли. Это кинетическая составляющая внутренней энергии молекул.
Для двух-атомного водорода атомы в молекуле попарно связаны друг с другом типа гантели. Можно предположить, что кроме движения относительно трех осей координат молекула водорода может вращаться подобно гантели относительно двух осей перпендикулярных двум плоскостям относительно продольной оси «гантели» (см. рис.2.2.1).

Рис. 2.2 Схематическое изображение молекулы водорода.
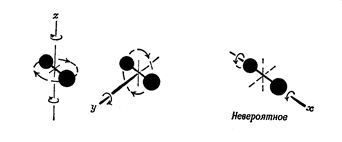
Рис.2.3. Степени свободы движения в молекуле
При высоких температурах свой вклад для двух-атомных молекул вносит колебательная энергия атомов в молекуле-гантели вдоль оси «гантели» (см. Рис.2.4).
Рис.2.4 Колебательные движения в молекуле.
Иллюстрацией изменения удельной теплоемкости водорода в зависимости от абсолютной температуры является Рис.2.5. По ординате рис. 2.5 отложено отношение молярной теплоемкости водорода при постоянном объеме к его газовой постоянной.
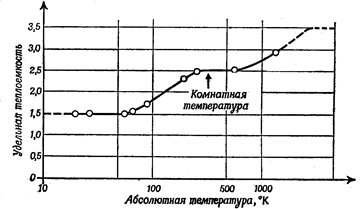
Рис.2.5 Изменение теплоемкости водорода от температуры.
Можно видеть, что при повышении температуры водорода энергия идет на следующие энергии движения:
Поступательное (3) всего 3 доли........ удельная теплоемкость 1,5
Поступательное (3) + враща-
тельное (2).............................................. удельная теплоемкость 2,5
Поступательное (3) + враща-
тельное (2) + колебательное (2).......... удельная теплоемкость 3,5
Эти ступеньки поведения удельной теплоемкости для каждого из ожидаемых значений были загадкой, пока не сообразили, что они получаются из ограничений на вращательную и колебательную энергии – ограничений квантового типа. Квантовые правила, возникшие при изучении другого явления - неожиданного поведения излучения, требуют, чтобы энергия на периодическое движение, такое, как колебания или вращения, воспринималась бы «порциями». Энергия каждой из таких «порций», или квантов, определяется правилом
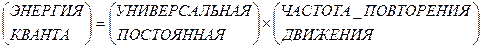
Сообщим молекуле один квант энергии вращения. Это заставит вращаться ее довольно быстро, ибо ее инерция вращения (момент инерции) довольно мала.
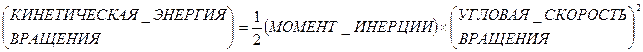 Но при быстром вращении молекулы кванты ее энергии должны стать большими. Поэтому молекулы поглощают энергию либо большими квантами, либо не поглощают вовсе и не вращаются. При низких температурах средняя доля, положенная по закону равномерного распределения, оказывается гораздо меньше одного кванта, так что вращаться могут лишь немногие молекулы. Частота колебаний атомов в молекуле очень высокая, поэтому практически ни одна молекула не может колебаться, пока газ не нагрет до очень высоких температур, что и иллюстрируется Рис.2.5
Но при быстром вращении молекулы кванты ее энергии должны стать большими. Поэтому молекулы поглощают энергию либо большими квантами, либо не поглощают вовсе и не вращаются. При низких температурах средняя доля, положенная по закону равномерного распределения, оказывается гораздо меньше одного кванта, так что вращаться могут лишь немногие молекулы. Частота колебаний атомов в молекуле очень высокая, поэтому практически ни одна молекула не может колебаться, пока газ не нагрет до очень высоких температур, что и иллюстрируется Рис.2.5
Оказалось, что для одноатомных молекул газов число степеней свободы i=3, а для двухатомных i= 5. Так что:
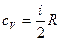
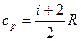
В заключение следует отметить очень важную особенность теплоемкостей идеального газа – теплоемкости ср и сV зависят т о л ь к о о т т е м п е р а т у р ы и не зависят от давления и объема.
Существуют понятия истинной и средней теплоемкости веществ. Все, что было сказано выше, относится к понятию истинной теплоемкости веществ. Истинная теплоемкость показывает количество теплоты, требуемое для нагревания массы вещества на один градус, т.е. истинная теплоемкость - есть частная производная сист = dQ/dT.
На практике чаще всего требуется определять количества теплоты для значительной разности температур. Тогда вводят осредненное по рассматриваемому диапазону температур значение теплоемкости с ср. Рассмотрим связь этих величин:
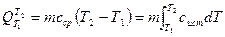 , откуда
, откуда 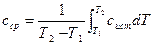
В приложении приведены уравнения для определения средней удельной теплоемкости наиболее часто применяемых газов в диапазоне температур от 0 до 15000С.
ІІІ. РЕАЛЬНЫЕ ГАЗЫ И ПАРЫ
Модель идеального газа хорошо описывает процессы, происходящие в реальных газах при сравнительно высоких температурах и низких давлениях (плотность газа мала, расстояния между отдельными молекулами несравнимо больше размеров самих молекул), внутренняя кинетическая энергия газа несравнимо больше внутренней потенциальной, и поэтому последней можно пренебречь. Другими словами, можно считать, что силы межмолекулярного сцепления отсутствуют. При этих же условиях можно пренебречь размерами самих молекул, так как они значительно меньше расстояния между ними.
Идеальный газ отвечает уравнению состояния Клапейрона-Менделеева
рVм=μRT (3.1)
где р – давление, кг/м2 или Па
Vм – объем моля, м3/кмоль
μ – молекулярный вес, кг/кмоль
R – газовая постоянная, кг·м/ кг·град или Дж/кг·град
Т – абсолютная температура, К
Однако для общего рассмотрения оказывается необходимым учитывать силы межмолекулярного сцепления и размеры молекул. Например, нельзя уравнениями идеального газа пользоваться для случаев фазовых переходов. Появляется необходимость учитывать особенности поведения реальных газов. Так для идеального и реальных газов можно рассмотреть поверхности состояния (рис. 3.1 и рис.3.2).
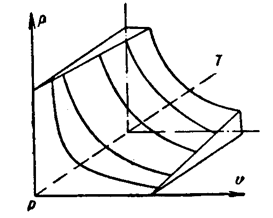
Рис.3.1 Поверхности состояния идеального газа.
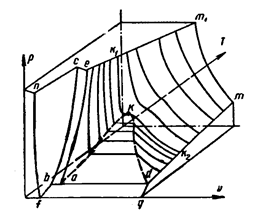
Рис. 3.2. Поверхности состояния реальных веществ.
В плоском изображении в координатах р-υ серия изотерм образует семейство равнобоких гипербол во всем диапазоне температур и давлений. В тех же координатах свойства реальных газов изображаются изотермами Эдрьюса (экспериментально найденными изотермами) в области влажного пара и некоторыми сложными кривыми в области перегретого пара (рис. 3.2). При повышении температур изотермы все больше приближаются к равнобоким гиперболам.
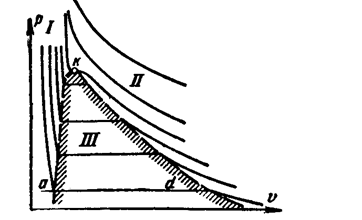
Рис.3.3. Изотермы Эндрьюса реальных газов.
Поверхность состояния реального газа (рис. 3.2) отражает общее качество для всех веществ – возможность существования в трех различных агрегатных состояниях: твердом, жидком и газообразном. Твердая фаза имеет место в условиях, сответствующих участку ncbf, жидкая фаза соответствует участку поверхности k1kae, и газовая фаза соответствует участку k1kdmm1. Помимо этого, на термодинамической поверхности можно выделить несколько характерных областей (так называемые гетерогенные области), где вещество распадается на две фазы, которые сосуществуют, а температура является только функцией давления.. Это области плавления, кипения и испарения твердых тел. Поэтому в каждой из этих областей уравнение поверхности состояния будет иметь вид f(T, p) = 0, что соответствует цилиндрическому участку поверхности. На рис.3.2 это цилиндрические участки akd (кипение), aecb (плавление) и fbdg (сублимация или возгонка). Три цилиндрические поверхности пересекаются в одной точке a, называемой тройной точкой. При давлении и температуре тройной точки вещество может существовать одновременно в трех фазовых состояниях.
Тип изображенной на рис. 3.2 термодинамической поверхности характерен для всех так называемых нормальных (неассоциирующих) веществ. Индивидуальные особенности того или иного вещества непосредственно отражаются на форме его термодинамической поверхности.
Очевидно, что отыскание универсальных уравнений состояния реальных веществ, учитывающих многообразные факторы, часто присущие только отдельным телам или отдельным состояниям, является чрезвычайно трудоемким и практически мало целесообразным, так как эти уравнения оказываются слишком громоздкими. В этом отношении более рационально определение групп «сходственных» веществ, для которых можно найти однотипное уравнение состояния. В отношении многих газообразных веществ такой группой является группа тел Амага, для которой уравнение состояния имеет вид:
[p+f1(υ)]f2(υ)=RT (3.2)
и группа тел, отвечающих наиболее общему уравнению М.П. Вукаловича – И.И. Новикова, учитывающего различную степень ассоциации молекул:
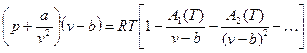 (3.3)
(3.3)
Характерным свойством группы тел Амага является независимость теплоемкости сυ от плотности. В общей сложности существует более 150 различных уравнений состояния реальных веществ; из них наиболее применяемыми для расчетов в области высоких давлений и низких температур являются следующие.
3.1. Уравнение ван-дер-Ваальса (1871 г.)
В своей диссертации «Непрерывность газобразных и жидких состояний» ван-дер-Ваальс предложил и обосновал следующее уравнение, относящееся к типу уравнений (3-2):
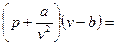 RT (3.4)
RT (3.4)
Оно получено в результате введения в уравнение состояния идеального газа поправок, учитывающих отличия реального газа от идеального. Силы межмолекулярного сцепления определяют поправку давления. Согласно данным Лапласа, силы сцепления стремятся удержать газ как единое целое, т.е. действуют от периферии объема, занимаемого газом, к его центру. Межмолекулярные силы взаимодействия «внутренних» частиц взаимно уравновешены в каждый момент времени и лишь молекулы, находящиеся у поверхности (у стенок) на расстоянии, меньшем радиуса действия сил сцепления, испытывают притяжение внутрь объема. Таким образом, направление действия этих сил совпадает с направлением действия сил внешнего даления р. На этом основании можно считать, что силы межмолекулярного сцепления увеличивают внешнее давление на некоторую величину рm, которую можно назвать внутренним или молекулярным давлением. Величина этого давления пропорциональна числу молекул, находящихся в данный момент времени у поверхности и пропорциональна числу молекул, находящихся внутри объема газа. Число молекул, расположенных у поверхности, так же как и число молекул во всем объеме, пропорционально плотности, и в результате суммарная сила межмолекулярных взаимодействий оказывается прямопропорциональной квадрату плотности газа или обратно пропорциональной квадрату удельного объема. Ван-дер Вааль предложил считать, что эта сила
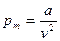 , где (3.5)
, где (3.5)
а – коэффициент, учитывающий природу вещества.
Поправка для объема определяется из следующих соображений. Наименьший объем, до которого можно сжать газ, обозначают через b. Этот объем назвали коволюм. Очевидно, что его надо вычесть из полного объема, занимаемого газом, т.к. он не изменяется при сжатии газа. Эти соображения и привели ван-дер-Вальса в 1971 году к уравнению (3.4). Значения констант a¸b и R в уравнении (3.4) приведены в таблице №3.1.
Построенная таким образом модель взаимодействия молекул, несмотря на значительное упрощение, оказалась плодотворной. Последующими исследованиями установлено, что молекулы не имеют определенных (жестких) размеров. Существующие между ними силы взаимного притяжения определяются существующими вокруг молекул элетрическими полями. Однако если молекулы сближены на такое расстояние, что их электронные оболочки приходят во взаимодействие, то появляется отталкивающая сила, величина которой быстро растет с уменьшением расстояния, и, наконец, превосходит силу притяжения. На рисунке 3.4 приведены кривые, характеризующие межмолекулярное силовое поле некоторых газов. Чем больше сила притяжения (больше величина «потенциальной ямы»), тем легче может быть сжижен газ (СО2) и, наоборот, для газов с весьма низкой критической температурой (трудносжимаемые газы Н2 и Не) характерна малая величина энергии притяжения.
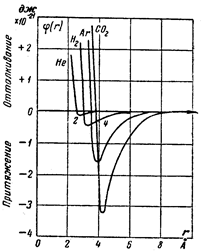
Рис.3.4 Влияние сил межмолекулярного сцепления для некоторых газов.
По шкале абсцисс отложено расстояние между молекулами в ангстремах (10-8мм)
Уточнения, определяемые реальной структурой молекулярных взаимодействий, приводят к уравнению состояния вида
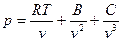 ....... (3.6)
....... (3.6)
где В, С и другие – вириальные коэффициенты, определяемые на основе эксперимента.
Такое разложение было получено Камерлинг – Оннесом.
3.1.1 Уравнение Д. Бертло (1903 г.)
Получено как результат уточнения уравнения ван-дер Ваальса. Бертло предложил
считать, что а обратно пропорционально Т. Обычный вид уравнения Бертло:
pυ=RT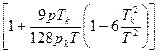 (3.7)
(3.7)
или 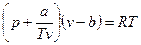 (3.8)
(3.8)
3.1.2. Уравнение Битти-Бриджмена (1927 г.)
Уравнение имеет вид:
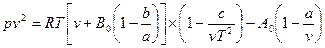 (3.9)
(3.9)
На основании соотношения (3.6) это уравнение записывают иногда следующим образом:
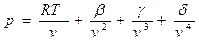 (3.10)
(3.10)
где
β= RTB0 - A0 - R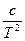 (3.11)
(3.11)
γ= - RTB0b + A0a - 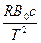 (3.12)
(3.12)
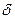 =
= 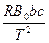 (3.13)
(3.13)
Уравнением Битти-Бриджмена можно пользоваться для вычисления значений р, υ, T в широком диапазоне температур и давлений. Точность уравнений весьма высока: например, для давлений до 100 атм расхождение с экспериментом составляет 0,16 – 0,18%. Значения коэффициентов, входящих в уравнение (3.9) Битти-Брижмена и (3.4) ван-дер-Ваальса, для различных газов даны в табл.№3.1, взятой из [8]. Таблица №3.1 Константы уравнений состояния Битти-Бриджмена и ван-дер-Ваальса
| Газы | Коэффициенты уравнения Битти-Бриджмена | Коэффициенты уравнения ван-дер - Ваальса | |||||
| А0 | а· 103 | В0· 103 | b· 103 | С· 10-4 | а, атм·л2 | b· 103, л | |
| Азот………………… | 1,3445 | 26,17 | 50,46 | - 6,91 | 4,2 | 1,347 | 38,6 |
| Аргон……………….. | 1,2907 | 23,28 | 39,31 | 0,0 | 5,99 | 1,35 | 32,2 |
| Водород…………….. | 0,1975 | -5,06 | 20,96 | -34,59 | 0,0504 | 0,245 | 26,6 |
| Воздух……………… | 1,3012 | 19,31 | 46,11 | -11,01 | 4,34 | 1,33 | 36,3 |
| Гелий………………. | 0,0216 | 59,84 | 14,00 | 0,0 | 0,004 | 0,033 | 23,2 |
| Кислород………….. | 1,4911 | 25,62 | 46,24 | 4,208 | 4,8 | 1,36 | 31,8 |
| Криптон……………. | 2,423 | 28,65 | 52,61 | 0,0 | 14,89 | 2,31 | 39,8 |
| Метан……………….. | 2,2769 | 18,55 | 55,87 | -15,87 | 12,83 | 2,20 | 42,8 |
| Неон………………… | 0,2125 | 29,96 | 20,6 | 0,0 | 0,101 | 2,16 | 62,0 |
| Углекислый газ…….. | 5,0065 | 71,32 | 104,76 | 72,35 | 66,00 | 3,6 | 42,8 |
| Окись углерода…….. | 1,3445 | 26,17 | 50,46 | -6,91 | 42,0 | 1,48 | 39,9 |
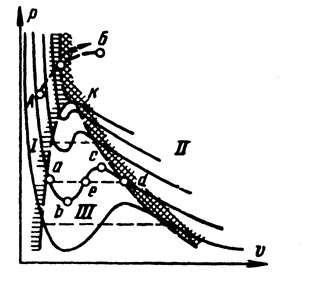
Рис.3.5 Реальные кривые изменения фазовых состояний веществ.
Принципиальной особенностью уравнения ван-дер-Ваальса является его справедливость как для газовой, так и для жидкой фаз, непосредственно вытекающая из непрерывности газообразных и жидких состояний и возможности рассмотрения молекулярной структуры этих фаз с единой точки зрения. Установленная ван-дер-Ваальсом непрерывность газообразных и жидких состояний означает возможность осуществления непрерывного перехода от газа к жидкости и в обратном направлении (переход a-d, рис.3.5). При этом существует определенная температура, называемая критической, при которой данное состояние вещества может быть достигнуто как исходя из газа, так и исходя из жидкости. Выше критической температуры вещество существует только в газовой фазе. Таким образом, газ может быть полностью или частично ожижен только при условии, если его температура ниже критической.
Критической точке соответствует и определенное значение давления рк. Вещество может существовать в двухфазном состоянии, если его температура и давление ниже критических. При давлении выше критического переход из газообразного состояния в жидкое возможен (разумеется, при Т<Tк, например, в результате процесса изобарного охлаждения), однако в любой точке такого процесса система будет оставаться однофазной.
При достаточно большом понижении температуры жидкость может перейти в твердую фазу. Весьма существенно, что этот переход всегда сопровождается скачком плотности; понятия «критической» точки перехода из твердой фазы в жидкую не существует. Это находит хорошее объяснение и с точки зрения молекулярной структуры твердого тела. Силы притяжения удерживают молекулы в положении около центров равновесия, образующих упорядоченную решетку. Тепловое движение (в этом случае колебания молекул около центров равновесия) оказывает несущественное влияние. При повышении температуры колебания становятся все значительнее и при достижении температуры плавления разрушают упорядоченную структуру, выводят тело из стабильного состояния. Такое разрушение или образование решетки и соответствует отмеченному здесь скачку плотности при взаимном переходе жидкой и твердой фаз.
Неустойчивые состояния возможны и в газовой, и в жидкой фазах. Для давлений ниже критического (рис. 3.5) уравнение ван-дер-Ваальса предполагает, что переход из жидкого состояния в газообразное осуществляется также непрерывно по изотерме abecd.
В действительности такая картина не наблюдается, и жидкость переходит в пар по изотерме ad. Если исходить из точки а, лежащей на так называемой нижней пограничной кривой, характеризующей состояние кипящей жидкости, то при осторожном ведении процесса можно получить начало участка ab. Это метастабильные (малоустойчивые) состояния перегретой жидкости. Так же можно получить и начало участка dc – метастабильные состояния пересыщенного или переохлажденного пара (точка d лежит на верхней пограничной кривой, характеризующей состояние насыщения). Участок bc практически осуществить невозможно. На участке ad изотермы ван-дер Ваальса и Эндрьюса раполагаются так, что площадки под изотермой (abea) и над нею (ecde) равны.
Так как критическая точка k представляет в конечом счете вырожденную изотерму ad, то касательная к критической изотерме в этой точке должна быть параллельной оси абсцисс, т.е. в этой точке производная
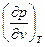 = 0 (3.14)
= 0 (3.14)
Одновременно точка k является точкой перегиба изотермы и поэтому производная
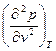 =0 (3.15)
=0 (3.15)
Критическая изотерма и пограничные кривые делят координатную плоскость на три области (Рис.3.5): I –область жидких состояний, II – область перегретого пара, III – область двухфазных состояний.
3.1.3 Критические параметры и константы уравнения ван-дер-Ваальса.
Из уравнения ван-дер-Ваальса
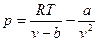 (3.16)
(3.16)
находим:
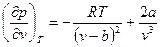 (3.17)
(3.17)
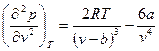 (3.18)
(3.18)
Приравнивая первую и вторую производные нулю, получим два уравнения. В результате решения системы уравнений (3.16) – (3.18) после простых преобразований найдем следующие значения параметров в критической точке:
р = рк = 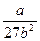 ü
ü
υ = υк = 3b ý (3.19)
Т = Тк = 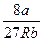 þ
þ
Из этих соотношений могут быть найдены константы уравнения ван-дер-Ваальса:
a = 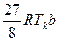 ; а = 27 рк b2 (3.20)
; а = 27 рк b2 (3.20)
b = 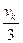 ; b =
; b = 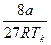 (3.21)
(3.21)
Уравнение ван-дер - Ваальса третьей степени и имеет три корня: υ1 , υ2 и υ3. В общем случае два из этих корней мнимые. Все три корня действительны только при р<рк и Т<Тк.
В этом случае, как пример, значения корней на рис. 3.5 представлены точками a, e и d.
Для критической точки υ1 = υ2 = υ3 = υк и тогда
(υ – υ1)(υ – υ2)(υ – υ3) == (υ – υк)3 = 0 (3.22)
Или
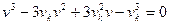 (3.23)
(3.23)
Сравнивая последнее уравнение с уравнением ван-дер- Ваальса в виде:
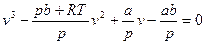 (3.24)
(3.24)
Заключаем
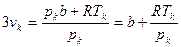
3υк2=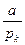 ;
; 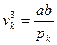 , откуда могут быть найдены и соотношения (3.20) – (3.21)
, откуда могут быть найдены и соотношения (3.20) – (3.21)
3.1.4. Приведенное уравнение ван-дер-Ваальса. Закон соответственных состояний.
Однозначная связь коэффициентов a, b и R с pк, υк и Tк позволяет исключить их из уравнения состояния. Подставим в уравнение ван- дер - Ваальса полученные значения констант из зависимостей (3.20) и (3.21). Тогда
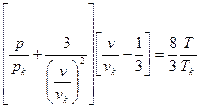 (3.25)
(3.25)
Принимая
р/рк = π – приведенное давление;
υ/υк = φ - приведенный объем;
Т/Тк = τ - приведенная температура,
запишем уравнение (3.25) в следующей форме:
(π + 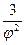 )(3φ -1) = 8τ (3.26)
)(3φ -1) = 8τ (3.26)
Уравнение (3.26) называют приведенным уравнением ван-дер-Ваальса. График этого уравнения называется приведенной диаграммой ван-дер-Ваальса (рис.3.6).
Приведенное уравнение и диаграмма в приведенных координатах одни и те же для любого вещества, что позволяет сформулировать весьма важный закон термодинамического подобия – закон соответственных состояний. Два или несколько различных веществ с двумя равными одноименными параметрами находятся в соответственных состояниях. При этом, несмотря на вообще присущие телам различия, их термодинамические поверхности совпадают. Более точным по отношению к какой-либо группе сходственных (подобных) веществ является экспериментальный закон соответственных состояний. В этом случае связи между приведенными величинами, найденные по опытным данным для одного вещества, могут быть распространены на другое. В частности, таким путем были предсказаны некоторые параметры гелия до его сжижения. При этом Камерлинг-Оннес исходил из данных, полученных для водорода.
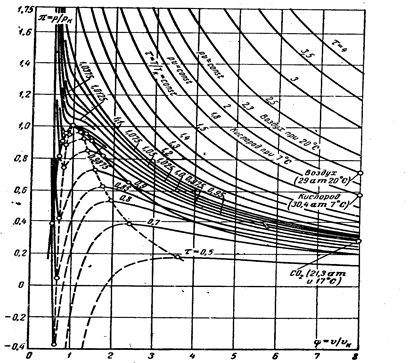
Рис.3.6. Приведенная диаграмма состояния веществ по ван-дер Ваальсу.
Конечно ожидать идеального соответствия данной диаграмме всех веществ не приходится, но, если уточнить исходные уравнения ван-дер-Ваальса по экспериментальным данным, то уточненная приведенная диаграмма позволяет сократить затраты на проведение экспериментов с каждым конкретным веществом.
3.2 ОБЩИЕ СООТНОШЕНИЯ ТЕРМОДИНАМИКИ.
3.2.1 Термодинамические потенциалы
Приведем основные соотношения для термомеханической системы, т.е. системы, обменивающейся с окружающей средой энергией в форме работы и тепла. Выбор независимых переменных для исследования такой системы определяется в каждом конкретном случае в зависимости от типа и характера изучаемого процесса. В общем случае для термомеханической системы с двумя степенями свободы независимые переменные можно выбирать из следующего ряда параметров:
р, υ; T, S; S, υ; S, р; T, υ; T, р
Первые две совокупности переменных относятся к одной степени свободы (первая – к «механической», а вторая - к «термической»), т.е. являются канонически сопряженными. Поэтому может быть образовано четыре равноценных зависимых переменных, являющихся функциями указанных параметров, причем каждая из них должна однозначно определять всю совокупность внешних воздействий, испытываемых системой. Эти четыре зависимые переменные для термомеханической системы – широко применяемые термодинамические функции или, как еще их часто называют, термодинамические потенциалы:
(U) u = f1(S;υ) – внутренняя энергия;
(I) i = f2 (S;p) = u + pυ - энтальпия;
(F) f = f3 (T; υ) = u – TS - свободная энергия;
(Φ) φ = f4(T;p) = I – TS = u + pυ – TS – свободная энтальпия, или термодинамический
потенциал Гиббса
Здесь S - энтропия; υ – удельный объем; р- давление; Т- температура
Математическая запись для этих функций:
du = TdS - рdυ; (3.27)
di = du + рdυ + υdр = TdS + υdр; (3.28)
df = du – TdS – SdT = -SdT- рdυ; (3.29)
dφ = di –TdS – SdT = - SdT + υdр; (3.30)
Легко убедиться, что дифференцирование любой из этих функций по какому-нибудь
параметру (независимой переменной) дает канонически сопряженный, например:
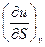 =T;
=T; 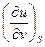 = - р;
= - р; 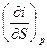 = T и т.д.
= T и т.д.
Как известно, в механике работа в поле сил не зависит от пути и определяется только разностью потенциалов в начальной и конечной точках пути. По аналогии, введенные термодинамические функции u, i, f и φ, однозначно определяющие всю совокупность внешних воздействий на систему в условиях равновесного взаимодействия независимо от пути (характера) процесса и дающие при дифференцировании сопряженные переменные, получили название термодинамических потенциалов.
Поскольку выражения du, di, df и dφ являются полными дифференциалами этих функций, то в соответствии со свойством уравнений в полных дифференциалах накрест взятые производные должны быть равны. Полученные таким образом дифференциальные соотношения термодинамики (соотношения Максвелла) устанавливают количественные связи между различными процессами и имеют большое значение в развитии термодинамического метода и исследовании различных систем. В таблице №3.2 приводятся перечисленные выше термодинамические функции.
Таблица №3.2
Таблица термодинамических функций.
| Термодинамические функции (потенциалы) | Независимые переменные | Сопряженные переменные | Дифференциальные термодинамические соотношения |
| U, u | υ,S du = TdS -рdυ | Т= р = - | 1-е уравнение Максвелла 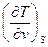 = - = - 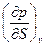 = = 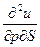 |
| I, i | р, S ∂i = TdS + υdр | Т= υ = 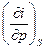 | 2-е уравнение Максвелла 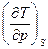 = = 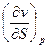 = = 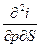 |
| F, f | υ, Т df = - SdT - рdυ | S = - 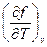 р = - р = - 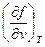 | 3-е уравнение Максвелла 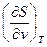 = =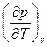 = =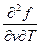 |
| Ф, φ | р, T dφ = - SdT + υdр | S = - 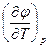 υ = υ = 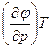 | 4-е уравнение Максвелла - 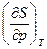 = =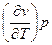 = =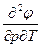 |
Приведенные выражения термодинамических потенциалов и коэффициентов в
дифференциальной форме позволяют получать частные решения для отдельных типов процессов:
изотермического – Т= const;
изобарного – р = const;
изохорного - υ = const;
адиабатного – dq = 0, которые нам понадобятся при рассмотрении циклов тепловых машин.
При помощи полученных дифференциальных соотношений важно найти выражения для определения различных параметров системы и термодинамических функций в зависимости от сочетания независимых переменных: Т и р; Т и v. Предварительно рассмотрим выражение, определяющее соотношение количества тепла в термодинамике и калориметрии:

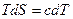 (3.31)
(3.31)
где с – теплоемкость тела. Как было отмечено выше, для газа теплоемкость зависит от характера протекающего процесса. В частности, широко распространены значения теплоемкости при р = const и v = const. В том случае из (43) находим:
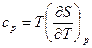 и
и 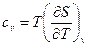 (3.32)
(3.32)
или 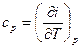 и
и 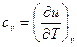 (3.32а)
(3.32а)
3.2.2. Физическая интерпретация термодинамических потенциалов.
Понятие функции внутренней энергии U непосредственно следует из закона сохранения и превращения энергии. Для любой термодинамической системы существует функция U, которая возрастает или убывает на величину подведенных или отнятых тепла и работы. При этом для систем, к которым тепло и работа подводятся или отнимаются в результате равновесных (квазистатических) процессов изменение функции U может быть однозначно определено через параметры самой системы.
Физическое содержание функции энтальпии i заключается в том, что для стационарной термодинамической системы или какого-либо рабочего процесса она однозначно определяет поток энергии. При использовании этой функции уравнения так называемых тепловых балансов для сложных систем становятся весьма простыми. В частных случаях:
а) когда поток энергии сохраняется, i = 0; такой процесс, известный под названием процесса дросселирования, характеризуется постоянством функции энтальпии и позволяет достаточно просто оценить понижение температуры предварительно сжатого вещества при проходе через дроссель (это явление используют при ожижении газов и в холодильных машинах);
б) когда давление остается постоянным (dp = 0), изменение энтальпии численно равно количеству подведенного или отведенного тепла. Это явление используют при рассмотрении циклов тепловых машин с подводом тепла при р = const.
Функция свободной энергии f, предложенная Гельмгольцем, определяет максимальную работу, которую может произвести система над окружающей средой или, наоборот, минимальную работу, которую надо сообщить системе, чтобы перевести ее из одного заданного состояния в другое при условии, что температура начального и конечного состояний системы равны температуре окружающей среды, и теплообмен с внешней средой осуществляется обратимо. Простейшим подобным переходом является изотермический процесс. В этом случае
df = - SdT – рdυ = - рdυ = dL, (3.33)
т.е. свободная энергия – это та часть внутренней энергии, которая в изотермическом обратимом процессе переходит в работу. При этом U f = TS является «связанной» энергией, в связи с необходимостью осуществления теплообмена, и не может быть превращена в работу. Кроме того, при изучении физико-химических процессов свободная энергия является также мерой химического сродства.
Функция свободной энтальпии (В.Гиббс), часто называемая также просто термодинамическим потенциалом или полным термодинамическим потенциалом (Дюгем), определяет величину максимальной работы в изобарно-изотермной системе. Максимальная работа, отдаваемая неоднородной системой в окружающую среду, в результате обратимого изобарно – изотермного процесса равна убыли свободной энтальпии. Кроме того, равенство свободных энтальпий в такой системе означает наличие фазового равновесия.
Все эти термодинамические функции путем простых преобразований можно привести к понятию внутренней энергии и определить с их помощью числовое значение последней. Поэтому в более общей физической трактовке этого вопроса их следует рассматривать как функции, объективно существующие для любой термодинамической системы и однозначно определяющие всю совокупность внешних воздействий, испытываемых системой, через различные независимые переменные, причем при соответствующем выборе независимых переменных каждая из этих функций обладает свойствами потенциальной функции.
3.2.3 ТЕРМОДИНАМИЧЕСКИЕ ДИАГРАММЫ
СОСТОЯНИЯ РЕАЛЬНЫХВЕЩЕСТВ.
Термодинамика позволяет производить расчеты состояния тел в широком диапазоне параметров состояния, с учетом возможного перехода из одного фазового состояния в другое. Расчетные зависимости кинетической теории позволяют достоверно оценивать изменение и взаимную связь параметров состояния в паровой (газовой) фазе. Современное развитие техники требует обеспечения возможности расчетного определения параметров в более широком диапазоне их изменения. Это в первую очередь относится к криогенным процессам, т.е. процессам, производимым при высоких давлениях и низких значениях температур, где, как показано выше, расчетные методы имеют ограничение из-за неизбежности снижения точности расчетов.
Еще до внедрения в расчетные работы высоко-эффективной вычислительной техники для производства с инженерной точностью расчетов широкое распространение нашли термодинамические диаграммы состояния веществ, построенные на основании экспериментальных исследований.
Для лучшего понимания особенностей таких диаграмм и методов их использования необходимо знать способы их построения и допущения, принимаемые при этом. Особую ценность диаграммы состояния имеют в области фазовых переходов.
Рассмотрим для примера типовую диаграмму фазовых переходов.
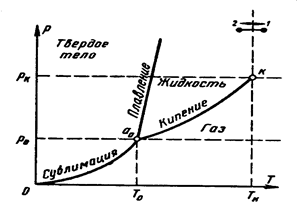
Рис. 3.7. Иллюстрация понятия тройной точки
а – тройная точка (проекция линии a-d рис.3.2 и 3.5), к – критическая точка
На рис.3.2 была приведена поверхность состояния реального рабочего тела в координатах p, υ и T. Ее можно рассматривать как фазовую диаграмму. Имеются характерные точки, определяющие фазовое состояние веществ, так называемые критические и тройные точки. Понятие тройной и критической точек ясно из Рис.3.7 и Рис.3.3. Критическая точка k определяет особенность перехода на границе газ-жидкость и будет рассмотрена ниже. При давлениях выше критического газообразная фаза переходит в жидкую и обратно без скачка плотности (процесс I-II). Кривая же плавления не имеет такого ограничения вплоть до очень высоких изученных уровней давлений.
Таблица №3.3

