 2015-04-23
2015-04-23 8754
8754Гидроксильная группа относится к числу групп, активирующих электрофильное замещение в ароматическом кольце и направляющих заместитель в орто - и пара -положения. Фенолы вступают практически во все типичные реакции электрофильного замещения как с сильными, так и со слабыми электрофильными агентами.
Галогенирование
Галогенирование фенолов не требует катализа кислотами Льюиса и легко осуществляется под действием молекулярного галогена. Галогенирование фенола молекулярным бромом или хлором в полярной среде практически невозможно остановить на стадии моногалогенирования, поскольку реагирующей частицей здесь является фенолят-ион, который содержит очень сильную активирующую группу –O-. Скорость галогенирования фенолят-иона по крайней мере в тысячу раз выше, чем фенола. Галогензамещенный фенол является более сильной кислотой, чем сам фенол, что облегчает введение второго и третьего атома галогена в орто - и пара- положения.
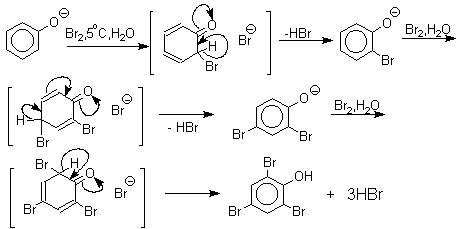
Бромирование фенола в воде приводит к образованию нерастворимого 2,4,6-трибромфенола. Эта реакция настолько чувствительна, что позволяет обнаружить фенол в концентрации 10-5М в водном растворе. 2,4,6-Трибромфенол взаимодействует еще с одним молем брома с образованием 2,4,4,6-тетрабромциклогекса-2,5-диенона, окрашенного в желтый цвет.
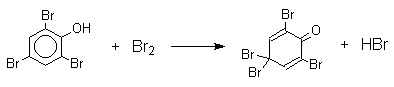
При обработке продукта этой реакции раствором гидросульфита натрия или другого слабого восстановителя он легко превращается в исходный 2,4,6-трибромфенол.
При бромировании фенола в растворе бромистоводородной кислоты диссоциация полностью подавляется и галогенированию подвергается сам фенол. При этом в зависимости от условий и количества галогена может быть получен п-бромфенол или 2,4-дибромфенол.
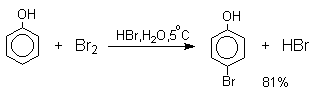
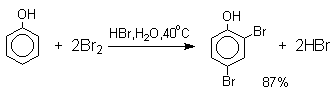
Аналогичным образом протекает хлорирование фенола хлором в соляной кислоте, но здесь получается значительное количество о-хлорфенола. Моногалогензамещенные производные фенолов удобно получать при галогенировании в неполярной среде, что также исключает диссоциацию фенолов.
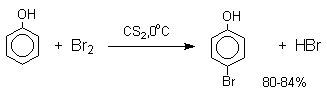
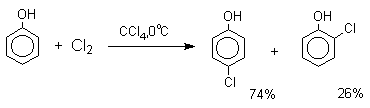
В качестве галогенирующего агента кроме самих галогенов можно использовать комплексы галогенов с диоксаном, ДМФА.
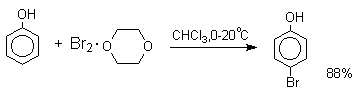
Во всех случаях соотношение пара / орто- изомеров при бромировании и иодировании значительно выше, чем при хлорировании.
Нитрование
Нитрование фенолов разбавленной 20-25%-ной азотной кислотой приводит к получению смеси орто- и пара-нитрофенолов.
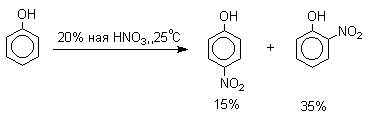
Даже в таких мягких условиях нитрование сопровождается окислением фенола и этот процесс становится доминирующим, если для нитрования использовать концентрированную азотную кислоту. Поэтому для получения 2,4,6-тринитрофенола (пикриновой кислоты) используют видоизмененный способ нитрования. Фенол первоначально сульфируют до 4-гидрокси-1,3-бензолдисульфокислоты, а затем нитруют азотной кислотой.
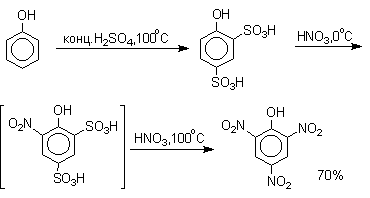
Вторая стадия по существу представляет собой электрофильное ипсо-замещение сульфогруппы на нитрогруппу.
Для нитрования фенолов в качестве нитрующего агента кроме азотной кислоты можно использовать ацетилнитрат и N2O4. Эти реагенты способствуют преимущественному нитрованию в орто -положение к гидроксильной группе.
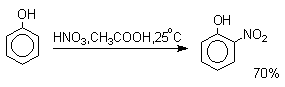
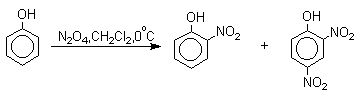
Сульфирование
Моносульфирование фенола серной кислотой приводит к образованию смеси орто- и пара-изомеров гидроксибензолсульфоксилоты. При 20оС в реакционной смеси содержится 49% орто-изомера и 51% пара-изомера, тогда как при 120оС доля пара-изомера возрастает до 96%.
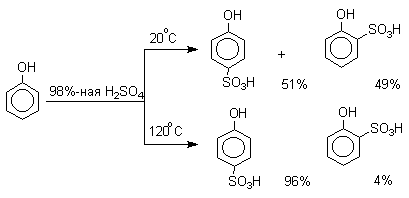
Изменение в соотношении продуктов сульфирования обусловлено обратимостью реакций, когда в равновесии преобладает термодинамически более стабильный пара -изомер. Сульфирование в орто -положение протекает с большей скоростью, но орто -гидроксибензолсульфокислота легко гидролизуется на исходные реагенты в отличие от пара -изомера, для которого скорость гидролиза мала.
Нитрозирование
Нитрозирование фенолов осуществляется с помощью азотистой кислоты в воде или уксусной кислоте. Эта реакция отличается очень высокой региоселективностью и замещение идет в пара -положение к гидроксильной группе. Типичное распределение орто - и пара -изомеров при нитрозировании можно проиллюстрировать на примере самого фенола.
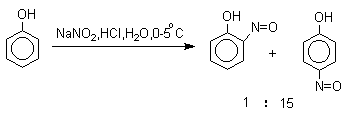
Алкилирование и ацилирование по Фриделю-Крафтсу
Так как фенолы взаимодействуют с галогенидами алюминия и другими кислотами Льюиса с образованием солей типа ArOAlCl2, прямое их алкилирование в условиях реакции Фриделя-Крафтса провести не удается. Фенолы алкилируют алкенами и спиртами в условиях кислотного катализа. В качестве катализаторов предпочитают использовать серную, фтористоводородную, фосфорную кислоты или катиониты КУ-2 и другие катионообменные смолы. Таким образом, из крезола и изобутилена в промышленности получают пространственно затрудненный фенол - 2,6-ди- трет- бутил-4-метилфенол (ионол), который широко применяется для стабилизации полимеров.
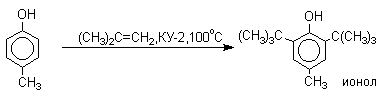
Аналогично из фенола и изопропилового спирта получается 2,4,6-триизопропилфенол.
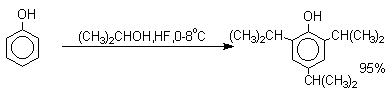
Ацилирование фенолов в классических условиях реакции Фриделя-Крафтса комплексом ацилгалогенида и хлорида алюминия приводит к неудовлетворительным результатам, так как ацилированию подвергается гидроксильная группа фенола. Более эффективна такая модификация этого метода, когда в качестве ацилирующего агента используется комплекс карбоновой кислоты и трехфтористого бора. Ацильная группа при этом вводится практически исключительно в пара -положение бензольного кольца. Так, например, фенол при взаимодействии с комплексом уксусной кислоты и BF3 дает пара -гидроксиацетофенон с 95%-ным выходом.
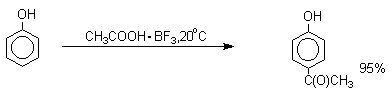
Наиболее общий метод получения гидроксикетонов ароматического ряда основан на перегруппировке Фриса. Ариловые эфиры карбоновых кислот при нагревании с AlCl3 или AlBr3перегруппировываются в изомерные орто - и пара -гидроксикетоны.
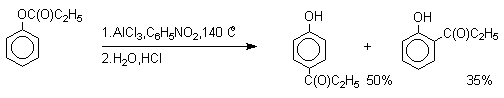
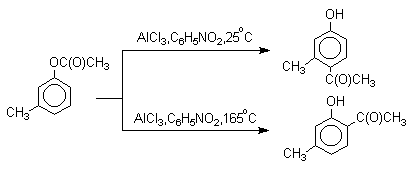
Соотношение орто- и пара- изомеров зависит главным образом от температуры и растворителя. В более жестких условиях преобладает орто -гидроксикетон, а при 20-25оС - пара -гидроксикетон.
Механизм перегруппировки Фриса, по-видимому, заключается в межмолекулярном ацилировании орто- или пара- положения бензольного кольца арилового эфира комплексом второй молекулы сложного эфира и AlCl3 с образованием ацильного производного гидроксикетона и фенола.
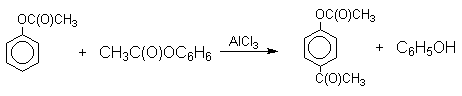
Перегруппировка завершается межмолекулярным переносом ацильной группы к фенолу.
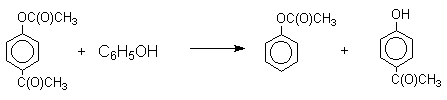
Формилирование
Формилирование – это введение группы СНО (см. лек.35). Синтетически наиболее важными являются формилирование фенолов по Вильсмейеру-Хааку и Реймеру-Тиману.
Реакция Вильсмейера-Хаака
N-Алкиламиды муравьиной кислоты - N,N-диметилформамид (ДМФА) и N-метилформамид в присутствии хлорокиси фосфора являются региоселективными формилирующими агентами. С помощью этих реагентов альдегидная группа вводится в пара-положение по отношению к ОН -группе. Эту реакцию можно также рассматривать как ацилирование, где роль катализатора (кислоты Льюиса) выполняют хлорокись фосфора (POCl3). Наиболее эффективна система ДМФА-POCl3, в которой ДМФА служит и реагентом, и растворителем. Электрофильным агентом в реакции Вильсмейера-Хаака является иминиевая соль, которая образуется при взаимодействии ДМФА и хлорокиси фосфора.
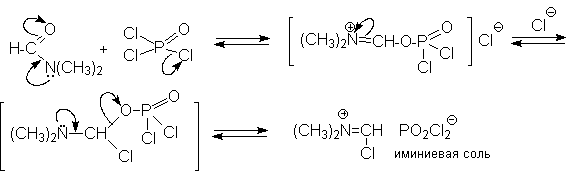
Иминиевая соль при необходимости может быть выделена в индивидуальном виде, однако обычно ее не выделяют и используют непосредственно после ее образования.
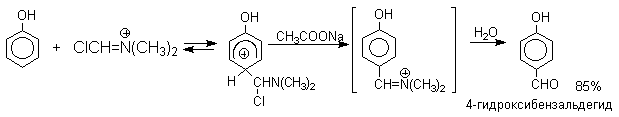
Реакция Вильсмейера-Хаака чрезвычайно проста в экспериментальном отношении и обеспечивает очень высокие выходы ароматических гидроксиальдегидов.
Реакция Реймера-Тимана
Формилирование фенолов по Реймеру-Тиману достигается при нагревании смеси фенола и большого избытка хлороформа с водным раствором гидроксида натрия при 50-70оС. Выходы альдегидов обычно невелики и редко превышают 30%, однако метод исключительно прост и доступен в практическом отношении. Главное достоинство реакции Реймера-Тимана заключается в преимущественном образовании орто-, а не пара-и зомеров, как это имеет место в реакции Вильсмейера-Хаака.
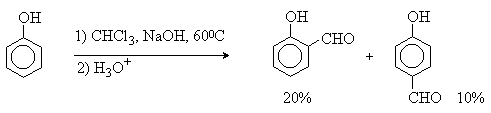
Механизм включает образование дихлоркарбена как интермедиата. Дихлоркарбен:CCl2 выполняет роль электрофильного агента по отношению к феноксид-иону, образующемуся в щелочной среде. Предполагаемый механизм реакции Реймера-Тимана может быть представлен следующей последовательностью превращений:
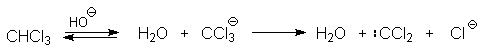
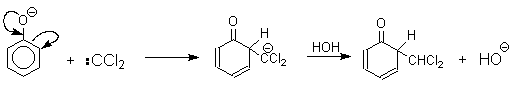
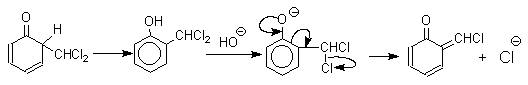
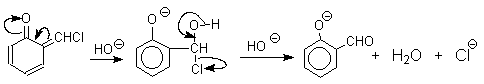
Реакция протекает только в сильно щелочной среде при наличии фенольного гидроксила, тогда как простые эфиры фенолов и диалкиланилины не формилируются в этих условиях.
Карбоксилирование феноксид-ионов (реакция Кольбе)
Будучи сильными нуклеофилами феноксид-анионы способны взаимодействовать с таким слабым электрофильным реагентом как оксид углерода (IV). При нагревании сухих фенолятов натрия или лития с СО2 при повышенном давлении, образуются натриевые или литиевые соли салициловой кислоты.
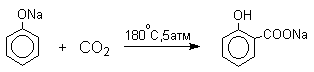
В аналогичных условиях из фенолятов калия, рубидия и цезия получаются только соли пара -гидроксибензойной кислоты.
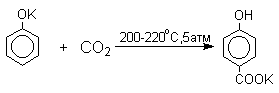
Такое различие в направлении карбоксилирования Na- и К-солей фенола принято объяснять различием в хелатообразовании этих двух катионов с атомом кислорода CO2 в переходном состоянии реакции, приводящем к салициловой кислоте. Катионы натрия и, особенно, лития значительно более эффективны по сравнению с катионом калия в способности к образованию координационной связи с атомом кислорода.
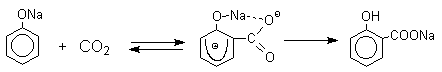
Предполагается, что для фенолятов калия, рубидия и цезия электрофильная атака осуществляется исключительно в пара -положение без какой-либо координации катиона по атому кислорода.
В отличие от одноатомных фенолов двухатомные и трехатомные фенолы карбоксилируются в более мягких условиях. Так, резорцин карбоксилируется при пропускании СО2 в водный раствор его дикалиевой соли при 50оС. При этом образуется 2,4-дигидроксибензойная кислота.
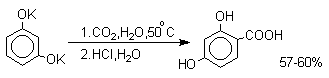
Конденсация с альдегидами и кетонами
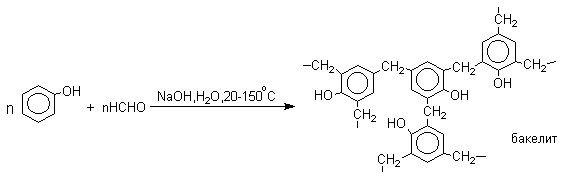
Фенолы реагируют с формальдегидом в водном растворе в присутствии основания с образованием полимерного продукта, получившего название феноло-формальдегидной смолы, карболита или бакелита.
Взаимодействие феноксид-иона с формальдегидом напоминает альдольную конденсацию с той лишь разницей, что роль нуклеофильного агента вместо енолят-иона выполняет амбидентный феноксид-ион, а карбонильной компонентой является формальдегид.
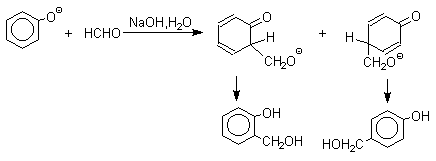
Подобно альдолям, орто - и пара -изомеры гидроксиметилфенола подвергаются дегидратации с образованием хинонметидов - соединений, родственных орто - и пара -хинонам.
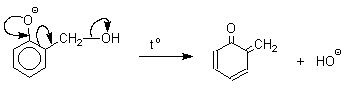
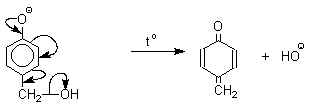
Последующее присоединение феноксид-иона к хинонметиду представляет собой присоединение амбидентного аниона к a,b -непредельному кетону по Михаэлю.
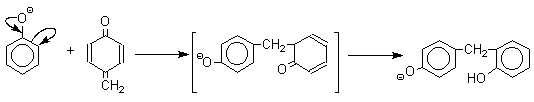
В результате дальнейшей поликонденсации в орто - и пара -положение к гидроксигруппе фенола получается трехмерная структура конечного продукта - бакелита. Бакелит представляет собой прозрачную смолу, в которой линейные звенья связаны "поперечными" связями в пара -положениях.
Фенол конденсируется с ацетоном в кислой среде с образованием так называемого бисфенола А.
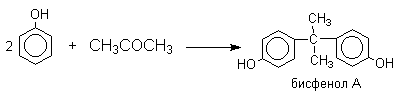
Получено много подобных продуктов конденсации фенолов с кетонами. Они находят применение в качестве антиоксидантов и мономеров для получения эпоксидных смол.

