2015-06-05
2015-06-05 779
77961). Химические свойства алкадиенов–1,3 (Сопряженных) Характерными реакциями всех алкадиенов являются реакции присоединения. Наиболее важными среди них являются: 1. Реакции гидрирования. Эти реакции имеют ступенчатый характер. Варьируя соотношение реагентов, может получать либо алкен, либо алкан в соответствии со схемой 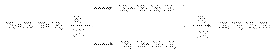 Катализаторами этих реакций являются Ni, Pt, Pd. Особенностью этих реакций является конкуренция 1,4– и 1,2–присоединения. Это связано с наличием двух реакционных центров у поверхностного интермедиата, образующегося в результате реализации механизма реакции
Катализаторами этих реакций являются Ni, Pt, Pd. Особенностью этих реакций является конкуренция 1,4– и 1,2–присоединения. Это связано с наличием двух реакционных центров у поверхностного интермедиата, образующегося в результате реализации механизма реакции 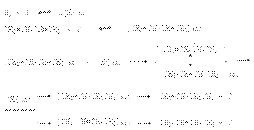 где S – свободная поверхность катализатора. 2. Реакции электрофильного присоединения а) Галогенирование При реализации этой реакции образуется смесь продуктов 1,2– и 1,4–присоединения, например:
где S – свободная поверхность катализатора. 2. Реакции электрофильного присоединения а) Галогенирование При реализации этой реакции образуется смесь продуктов 1,2– и 1,4–присоединения, например: 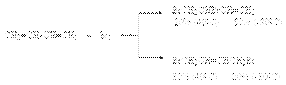 Образование продуктов конкурирующего галогенирования объясняется следующим механизмом:
Образование продуктов конкурирующего галогенирования объясняется следующим механизмом: 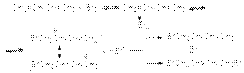 Можно видеть, что промежуточный карбкатион имеет два реакционных центра, что и обусловливает образование продуктов конкурентного галогенирования.
Можно видеть, что промежуточный карбкатион имеет два реакционных центра, что и обусловливает образование продуктов конкурентного галогенирования.
При углублении реакции образуются продукты исчерпывающего галогенирования: 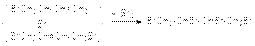 Изменение состава продуктов первичного галогенирования при изменении температуры обусловлено изменением характера факторов, определяющих соотношение продуктов. Так, в области высоких температур конкурирующие реакции быстро достигают положения равновесия, и состав продуктов будет определяться соотношением их равновесных концентраций. Таким образом, в области высоких температур осуществляется термодинамический контроль состава продуктов. В области низких температур из-за высокой экзотермичности каждой из конкурирующих реакций их равновесия существенно смещены в сторону продуктов, а скорости их слишком малы, чтобы заметно приблизиться к состоянию равновесия. Поэтому обе реакции протекают в условиях практической необратимости, когда состав продуктов определяется соотношением их скоростей. Это областькинетического контроля состава продуктов. Энергетический профиль реакции бромид-аниона с промежуточным карбкатионом иллюстрирует факторы, управляющие такого рода процессами.
Изменение состава продуктов первичного галогенирования при изменении температуры обусловлено изменением характера факторов, определяющих соотношение продуктов. Так, в области высоких температур конкурирующие реакции быстро достигают положения равновесия, и состав продуктов будет определяться соотношением их равновесных концентраций. Таким образом, в области высоких температур осуществляется термодинамический контроль состава продуктов. В области низких температур из-за высокой экзотермичности каждой из конкурирующих реакций их равновесия существенно смещены в сторону продуктов, а скорости их слишком малы, чтобы заметно приблизиться к состоянию равновесия. Поэтому обе реакции протекают в условиях практической необратимости, когда состав продуктов определяется соотношением их скоростей. Это областькинетического контроля состава продуктов. Энергетический профиль реакции бромид-аниона с промежуточным карбкатионом иллюстрирует факторы, управляющие такого рода процессами. 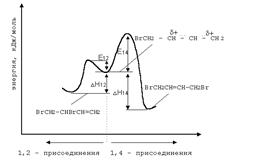 Можно видеть, что образование продукта 1,4–присоединения термодинамические более выгодно, т.к. теплота D Н1,4 – более электроотрицательна, чем D Н1,2. Поэтому в области термодинамического контроля предпочтительно образуется продукт 1,4–присоединения. В то же время энергетический барьер образования продукта 1,2–присоединения (Е1,2) ниже, чем энергетический барьер образования продукта 1,4–присоединения (Е1,4). Поскольку система легче преодолевает более низкий барьер, то в области кинетического контроля преобладающим будет продукт 1,2–присоединения. В целом вероятность термодинамического контроля возрастает с ростом температуры и времени реакции. Наоборот, вероятность кинетического контроля тем больше, чем ниже температура и время реакции. б) Реакции гидрогалогенирования. При реализации этих реакций образуются продукты конкурентного галогенирования:
Можно видеть, что образование продукта 1,4–присоединения термодинамические более выгодно, т.к. теплота D Н1,4 – более электроотрицательна, чем D Н1,2. Поэтому в области термодинамического контроля предпочтительно образуется продукт 1,4–присоединения. В то же время энергетический барьер образования продукта 1,2–присоединения (Е1,2) ниже, чем энергетический барьер образования продукта 1,4–присоединения (Е1,4). Поскольку система легче преодолевает более низкий барьер, то в области кинетического контроля преобладающим будет продукт 1,2–присоединения. В целом вероятность термодинамического контроля возрастает с ростом температуры и времени реакции. Наоборот, вероятность кинетического контроля тем больше, чем ниже температура и время реакции. б) Реакции гидрогалогенирования. При реализации этих реакций образуются продукты конкурентного галогенирования: 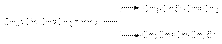 Механизм образования продуктов может быть представлен следующими стадиями:
Механизм образования продуктов может быть представлен следующими стадиями: 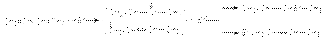 В области высоких температур состав продуктов определяется термодинамическим контролем и доминирующим продуктом является продукт 1,4–присоединения. В области низких температур состав продуктов определяется кинетическим контролем и основным продуктом является продукт 1,2–присоединения. Таким образом наблюдается полная аналогия между реакциями галогенирования и гидрогалогенирования 1,3-алкадиенов. в) Полимеризация Стехиометрически полимеризация сопряженных алкадиенов описывается следующими уравнениями
В области высоких температур состав продуктов определяется термодинамическим контролем и доминирующим продуктом является продукт 1,4–присоединения. В области низких температур состав продуктов определяется кинетическим контролем и основным продуктом является продукт 1,2–присоединения. Таким образом наблюдается полная аналогия между реакциями галогенирования и гидрогалогенирования 1,3-алкадиенов. в) Полимеризация Стехиометрически полимеризация сопряженных алкадиенов описывается следующими уравнениями 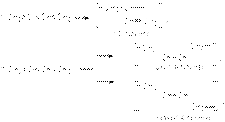 Полимеризация сопряженных алкадиенов имеет важное практическое значение для синтеза каучуков. Реакция может осуществляться по свободнорадикальному, карбанионному и карбкатионному механизмам в зависимости от типа инициирующей системы. В промышленной практике наиболее широко реализуется стереорегулярная полимеризация под действием катализаторов Циглера-Натта.
Полимеризация сопряженных алкадиенов имеет важное практическое значение для синтеза каучуков. Реакция может осуществляться по свободнорадикальному, карбанионному и карбкатионному механизмам в зависимости от типа инициирующей системы. В промышленной практике наиболее широко реализуется стереорегулярная полимеризация под действием катализаторов Циглера-Натта.
Механизм радикальной полимеризации может быть представлен на примере 1,4–присоединения. Инициирование 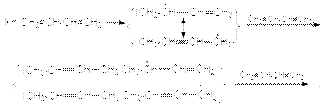 Таким образом, в результате актов многократного присоединения растущего радикала к алкадиену происходит рост цепи будущего полимера. Обрыв цепей осуществляется рекомбинацией или диспропорционированием макрорадикалов. Можно видеть, что растущие радикалы обладают двойственной реакционной способностью, что открывает возможность образования 1,4– и 1,2–полимера. Обычно при свободнорадикальной полимеризации доминирующим является 1,4–полимер с примесью 1,2–полимера, причем первый в основном состоит из транс-изомера. В качестве инициаторов этих реакций используют пероксиды или азобисизобутиронитрил. В присутствии металлического натрия осуществляется полимеризация по анион-радикальному механизму:
Таким образом, в результате актов многократного присоединения растущего радикала к алкадиену происходит рост цепи будущего полимера. Обрыв цепей осуществляется рекомбинацией или диспропорционированием макрорадикалов. Можно видеть, что растущие радикалы обладают двойственной реакционной способностью, что открывает возможность образования 1,4– и 1,2–полимера. Обычно при свободнорадикальной полимеризации доминирующим является 1,4–полимер с примесью 1,2–полимера, причем первый в основном состоит из транс-изомера. В качестве инициаторов этих реакций используют пероксиды или азобисизобутиронитрил. В присутствии металлического натрия осуществляется полимеризация по анион-радикальному механизму: 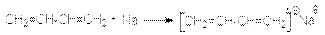 Обозначая
Обозначая 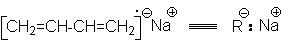 можно представить последующий путь полимеризации совокупностью стадий:
можно представить последующий путь полимеризации совокупностью стадий: 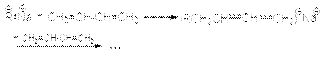 Полимеризация обычно протекает на поверхности металла, поэтому из-за стерических требований к реакции преимущественно образуется 1,2–полимер. Полимеризация по Циглеру-Натта приводит к образованию стереорегулярного каучука, причем в основном образуются цис-полиалкадиены. 3. Реакции диенового синтеза Алкадиены–1,3 могут присоединяться к двойной (или тройной) связи с образованием циклического продукта (циклоприсоединение [2+4]).
Полимеризация обычно протекает на поверхности металла, поэтому из-за стерических требований к реакции преимущественно образуется 1,2–полимер. Полимеризация по Циглеру-Натта приводит к образованию стереорегулярного каучука, причем в основном образуются цис-полиалкадиены. 3. Реакции диенового синтеза Алкадиены–1,3 могут присоединяться к двойной (или тройной) связи с образованием циклического продукта (циклоприсоединение [2+4]).  Такие реакции называются диеновым синтезом или реакцией Дильса-Альдера. Соединения, содержащие двойную или тройную связь и вступающие с 1,3–алкадиенами в реакцию диенового синтеза называются диенофилами. Реакционная способность диенофилов увеличивается при активировании их кратной связи электроноакцепторными группами. В реакции Дильса-Альдера происходит исчезновение трех старых p -связей и появление новых: одной p -связи и двух s -связей. Поскольку прочность s -связей существенно выше, чем прочность p -связей, реакция Дильса-Альдера экзотермична. Это обусловливает ее обратимость при высоких температурах. Условием успешного протекания реакции является такая геометрия реагентов, при которой оба конца диена ориентированы на углеродные атомы кратной связи диенофила. При этом происходит согласованный процесс образования новых и разрыва старых связей с синхронным перемещением всех p -электронов циклической системы
Такие реакции называются диеновым синтезом или реакцией Дильса-Альдера. Соединения, содержащие двойную или тройную связь и вступающие с 1,3–алкадиенами в реакцию диенового синтеза называются диенофилами. Реакционная способность диенофилов увеличивается при активировании их кратной связи электроноакцепторными группами. В реакции Дильса-Альдера происходит исчезновение трех старых p -связей и появление новых: одной p -связи и двух s -связей. Поскольку прочность s -связей существенно выше, чем прочность p -связей, реакция Дильса-Альдера экзотермична. Это обусловливает ее обратимость при высоких температурах. Условием успешного протекания реакции является такая геометрия реагентов, при которой оба конца диена ориентированы на углеродные атомы кратной связи диенофила. При этом происходит согласованный процесс образования новых и разрыва старых связей с синхронным перемещением всех p -электронов циклической системы  Поскольку диен может реагировать в s-цисоидной конформации, реакции некоторых диенов являются стерически запрещенными. Примером таких диенов являются приведенные ниже структуры
Поскольку диен может реагировать в s-цисоидной конформации, реакции некоторых диенов являются стерически запрещенными. Примером таких диенов являются приведенные ниже структуры 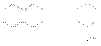 С точки зрения молекулярно-орбитального рассмотрения циклоприсоединение является согласованным, разрешенным по симметрии процессом. Из нижеприведенной молекулярно-орбитальной картины можно видеть, что симметрия ВЗМО бутадиена и НСМО этилена, а также ВЗМО этилена и НСМО бутадиена одинакова. В результате энергия возмущения велика и реакция циклизации может протекать как согласованный процесс.
С точки зрения молекулярно-орбитального рассмотрения циклоприсоединение является согласованным, разрешенным по симметрии процессом. Из нижеприведенной молекулярно-орбитальной картины можно видеть, что симметрия ВЗМО бутадиена и НСМО этилена, а также ВЗМО этилена и НСМО бутадиена одинакова. В результате энергия возмущения велика и реакция циклизации может протекать как согласованный процесс. 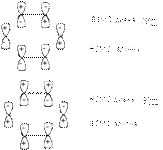 Реакция Дильса-Альдера высокостереоспецифична, что проявляется в сохранении конфигурации диена и диенофила в продукте циклизации. Это наглядно видно на примере цис– и транс–дизамещенных алкенов, используемых в качестве диенофилов:
Реакция Дильса-Альдера высокостереоспецифична, что проявляется в сохранении конфигурации диена и диенофила в продукте циклизации. Это наглядно видно на примере цис– и транс–дизамещенных алкенов, используемых в качестве диенофилов: 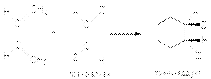
62). Каучук. Представления о строении натурального каучука. Синтетические каучуки. Механизм реакции полимеризации. Натуральный каучук представляет собой эластичную при низких температурах, пластичную и клейкую при более высоких температурах массу, которую получают при нагревании млечного сока растений – каучуконосов, таких как гевея. Первое практическое применение натурального каучука нашел Макинтош (1823) – при пропитывании тканей раствором каучука он получил водонепроницаемый материал. Наибольшее значение для широкого использования каучука имело открытие Гудьира (1839). Он обнаружил, что при обработке каучука серой или серосодержащими соединениями получается материал с превосходными механическими свойствами. Так была впервые получена резина. Сера, реагируя по аллильному водороду и двойной связи образует мостики, связывающие между собой полимерные цепи. 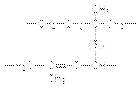 Этот процесс называется вулканизацией. При исследовании строения натурального каучука было показано, что он является полимером изопрена. Позже на основе рентгеноструктурного анализа было установлено, что натуральный каучук имеет строение цис-полиизопрена. В природе встречается также транс-полиизопрен, называемый гуттаперчей, который является твердым хрупким материалом и поэтому не находит широкого практического применения. В СССР впервые синтетический каучук был получен на основе бутадиена–1,3 при катализе Na или K (1932) – метод С.В. Лебедева. Промышленная партия стереорегулярного полиизопрена была выпущена у нас в стране в 1963 – так называемый каучук СКИ-3. По своим свойствам этот каучук не уступает натуральному. В 1956 году под руководством советского химика Долгоплоска был разработан метод стереорегулярной полимеризации 1,3–бутадиена и получения на его основе цис-полибутадиенового каучука (дивинилкаучук, СКД). По эластичности СКД не уступает СКИ-3, но превосходит его в устойчивости при низких температурах и износостойкости. Спектр практического использования каучуков существенно расширен за счет синтеза сополимеров бутадиена со стиролом, бутадиена с акрилонитрилом и др. Природный каучук Сбор латекса гевеи (Шри-Ланка) Высокомолекулярный углеводород (C5H8)n, цис-полимер изопрена; содержится в млечном соке (латексе) гевеи, кок-сагыза (многолетнего травянистого растения рода Одуванчик) и других каучуконосных растений. Растворим вуглеводородах и их производных (бензине, бензоле, хлороформе, сероуглероде и т. д.).В воде, спирте, ацетоненатуральный каучук практически не набухает и не растворяется. Уже при комнатной температуре натуральный каучук присоединяет кислород, происходит окислительная деструкция (старение каучука), при этом уменьшается его прочность и эластичность. При температуре выше 200 °C натуральный каучук разлагается с образованием низкомолекулярных углеводородов. При взаимодействии натурального каучука с серой, хлористой серой, органическими пероксидами (вулканизация) происходит соединение через атомы серы длинных макромолекулярных связей с образованием сетчатых структур. Это придает каучуку высокую эластичность в широком интервале температур. Натуральный каучук перерабатывают в резину. В сыром виде применяют не более 1 % добываемого натурального каучука (резиновый клей). Каучук открыт де ла Кондамином в Кито (Эквадор) в 1751 году. Более 60 % натурального каучука используют для изготовления автомобильных шин. В промышленных масштабах натуральный каучук производится в Индонезии, Малайзии, Вьетнаме и Таиланде. Синтетические каучуки Первым синтетическим каучуком, имевшим промышленное значение, был полибутадиеновый (дивиниловый) каучук, производившийся синтезом по методу С. В. Лебедева (получение из этилового спирта бутадиена с последующей анионной полимеризацией жидкого бутадиена в присутствии натрия). В 1932 году в Ярославле запущен завод СК-1, работающий на основе этого метода, который стал первым в мире заводом по производству синтетического каучука в промышленных масштабах. В Германии бутадиен-натриевый каучук нашёл довольно широкое применение под названием «Буна». Синтез каучуков стал значительно дешевле с изобретением катализаторов Циглера — Натта. Изопреновые каучуки — синтетические каучуки, получаемые полимеризацией изопрена в присутствии катализаторов — металлического лития, перекисных соединений. В отличие от других синтетических каучуков изопреновые каучуки, подобно натуральному каучуку, обладают высокой клейкостью и незначительно уступают ему в эластичности. В настоящее время большая часть производимых каучуков является бутадиен-стирольными или бутадиен-стирол-акрилонитрильными сополимерами. Каучуки с гетероатомами в качестве заместителей или имеющими их в своём составе часто характеризуются высокой стойкостью к действию растворителей, топлив и масел, устойчивостью к действию солнечного света, но обладают худшими механическими свойствами. Наиболее массовым в производстве и применении каучуками с гетерозаместителями являются хлоропреновые каучуки (неопрен) — полимеры 2-хлорбутадиена. В ограниченном масштабе производятся и используются тиоколы — полисульфидные каучуки, получаемые поликонденсацией дигалогеналканов (1,2-дихлорэтана, 1,2-дихлорпропана) и полисульфидов щелочных металлов. Основные типы синтетических каучуков: Изопреновый, Бутадиеновый, Бутадиен-метилстирольный, Бутилкаучук (изобутилен-изопреновый сополимер), Этилен-пропиленовый (этилен-пропиленовый сополимер), Бутадиен-нитрильный (бутадиен-акрилонитрильный сополимер), Хлоропреновый (поли-2-хлорбутадиен), Силоксановый, Фторкаучуки, Тиоколы Механизм полимеризации обычно включает в себя ряд связанных стадий: инициирование — зарождение активных центров полимеризации; рост (продолжение) цепи — процесс последовательного присоединения молекул мономеров к центрам; передача цепи — переход активного центра на другую молекулу; разветвление цепи — образование нескольких активных центров из одного; обрыв цепи — гибель активных центров. Инициация заключается в образовании активных частиц, способных начать реакцию роста. Активная частица (А*) присоединяется к молекуле мономера(М), образуется новая активная частица, к ней снова присоединяется молекула мономера, вновь образуется активная частица и т.д. + М + М + М; А* + М ——> АМ* ————> АММ *————> АМММ*————> ….; Механизм реакции определяется а) природой инициатора А* б) природой промежуточной частицы АМ*. Инициатор А* может быть радикалом или ионом - катионом, анионом, и также существует два принципиально различных механизма полимеризации: радикальный и ионный(катионный или анионный). Если промежуточная частица АМ* неустойчивая и время ее жизни короткое, то механизм реакции цепной. Если АМ* более устойчивая частица, время ее жизни достаточно большое, то механизм реакцииступенчатый. Радикальная полимеризация Радикальная полимеризация всегда протекает по цепному механизму. Не каждый мономер может участвовать в радикальной реакции полимеризации. Радикальный механизм полимеризации возможен для этилена, хлорвинила, акрилонитрила, винилацетата, метакрилата, метилметакрилата. Образование инициаторов реакции - свободных радикалов Начало реакции: образование свободных радикалов, инициирующих начало реакции. +R* + CН2 = C Н2 + CН2 = C Н2; CН2 = C Н2——> R-СН2 –СН2 * ————> R-СН2 –СН2 —СН2 –СН2*————>…; Свободные радикалы могут быть получены несколькими путями: а) с участием инициаторов б) фотолитическим путем в) окислительно-восстановительной реакцией Наиболее распространенными инициаторами являются перекиси органических соединений и азосоединения. Перекиси обычно присутствуют в смесях, которые используют для приготовления полимерных изделий в стоматологии (при пломбировании, протезировании). Под влияние УФ-облучения происходит разры химический связи и образуются свободные радикалы УФ R – O – O– R ———> 2 R – O• (перекись алкила УФ); С6 Н5 – C– O– O– C – С 6Н5 ———> С6 Н5 – C– O• + С 6Н5• + СО2
Этот процесс называется вулканизацией. При исследовании строения натурального каучука было показано, что он является полимером изопрена. Позже на основе рентгеноструктурного анализа было установлено, что натуральный каучук имеет строение цис-полиизопрена. В природе встречается также транс-полиизопрен, называемый гуттаперчей, который является твердым хрупким материалом и поэтому не находит широкого практического применения. В СССР впервые синтетический каучук был получен на основе бутадиена–1,3 при катализе Na или K (1932) – метод С.В. Лебедева. Промышленная партия стереорегулярного полиизопрена была выпущена у нас в стране в 1963 – так называемый каучук СКИ-3. По своим свойствам этот каучук не уступает натуральному. В 1956 году под руководством советского химика Долгоплоска был разработан метод стереорегулярной полимеризации 1,3–бутадиена и получения на его основе цис-полибутадиенового каучука (дивинилкаучук, СКД). По эластичности СКД не уступает СКИ-3, но превосходит его в устойчивости при низких температурах и износостойкости. Спектр практического использования каучуков существенно расширен за счет синтеза сополимеров бутадиена со стиролом, бутадиена с акрилонитрилом и др. Природный каучук Сбор латекса гевеи (Шри-Ланка) Высокомолекулярный углеводород (C5H8)n, цис-полимер изопрена; содержится в млечном соке (латексе) гевеи, кок-сагыза (многолетнего травянистого растения рода Одуванчик) и других каучуконосных растений. Растворим вуглеводородах и их производных (бензине, бензоле, хлороформе, сероуглероде и т. д.).В воде, спирте, ацетоненатуральный каучук практически не набухает и не растворяется. Уже при комнатной температуре натуральный каучук присоединяет кислород, происходит окислительная деструкция (старение каучука), при этом уменьшается его прочность и эластичность. При температуре выше 200 °C натуральный каучук разлагается с образованием низкомолекулярных углеводородов. При взаимодействии натурального каучука с серой, хлористой серой, органическими пероксидами (вулканизация) происходит соединение через атомы серы длинных макромолекулярных связей с образованием сетчатых структур. Это придает каучуку высокую эластичность в широком интервале температур. Натуральный каучук перерабатывают в резину. В сыром виде применяют не более 1 % добываемого натурального каучука (резиновый клей). Каучук открыт де ла Кондамином в Кито (Эквадор) в 1751 году. Более 60 % натурального каучука используют для изготовления автомобильных шин. В промышленных масштабах натуральный каучук производится в Индонезии, Малайзии, Вьетнаме и Таиланде. Синтетические каучуки Первым синтетическим каучуком, имевшим промышленное значение, был полибутадиеновый (дивиниловый) каучук, производившийся синтезом по методу С. В. Лебедева (получение из этилового спирта бутадиена с последующей анионной полимеризацией жидкого бутадиена в присутствии натрия). В 1932 году в Ярославле запущен завод СК-1, работающий на основе этого метода, который стал первым в мире заводом по производству синтетического каучука в промышленных масштабах. В Германии бутадиен-натриевый каучук нашёл довольно широкое применение под названием «Буна». Синтез каучуков стал значительно дешевле с изобретением катализаторов Циглера — Натта. Изопреновые каучуки — синтетические каучуки, получаемые полимеризацией изопрена в присутствии катализаторов — металлического лития, перекисных соединений. В отличие от других синтетических каучуков изопреновые каучуки, подобно натуральному каучуку, обладают высокой клейкостью и незначительно уступают ему в эластичности. В настоящее время большая часть производимых каучуков является бутадиен-стирольными или бутадиен-стирол-акрилонитрильными сополимерами. Каучуки с гетероатомами в качестве заместителей или имеющими их в своём составе часто характеризуются высокой стойкостью к действию растворителей, топлив и масел, устойчивостью к действию солнечного света, но обладают худшими механическими свойствами. Наиболее массовым в производстве и применении каучуками с гетерозаместителями являются хлоропреновые каучуки (неопрен) — полимеры 2-хлорбутадиена. В ограниченном масштабе производятся и используются тиоколы — полисульфидные каучуки, получаемые поликонденсацией дигалогеналканов (1,2-дихлорэтана, 1,2-дихлорпропана) и полисульфидов щелочных металлов. Основные типы синтетических каучуков: Изопреновый, Бутадиеновый, Бутадиен-метилстирольный, Бутилкаучук (изобутилен-изопреновый сополимер), Этилен-пропиленовый (этилен-пропиленовый сополимер), Бутадиен-нитрильный (бутадиен-акрилонитрильный сополимер), Хлоропреновый (поли-2-хлорбутадиен), Силоксановый, Фторкаучуки, Тиоколы Механизм полимеризации обычно включает в себя ряд связанных стадий: инициирование — зарождение активных центров полимеризации; рост (продолжение) цепи — процесс последовательного присоединения молекул мономеров к центрам; передача цепи — переход активного центра на другую молекулу; разветвление цепи — образование нескольких активных центров из одного; обрыв цепи — гибель активных центров. Инициация заключается в образовании активных частиц, способных начать реакцию роста. Активная частица (А*) присоединяется к молекуле мономера(М), образуется новая активная частица, к ней снова присоединяется молекула мономера, вновь образуется активная частица и т.д. + М + М + М; А* + М ——> АМ* ————> АММ *————> АМММ*————> ….; Механизм реакции определяется а) природой инициатора А* б) природой промежуточной частицы АМ*. Инициатор А* может быть радикалом или ионом - катионом, анионом, и также существует два принципиально различных механизма полимеризации: радикальный и ионный(катионный или анионный). Если промежуточная частица АМ* неустойчивая и время ее жизни короткое, то механизм реакции цепной. Если АМ* более устойчивая частица, время ее жизни достаточно большое, то механизм реакцииступенчатый. Радикальная полимеризация Радикальная полимеризация всегда протекает по цепному механизму. Не каждый мономер может участвовать в радикальной реакции полимеризации. Радикальный механизм полимеризации возможен для этилена, хлорвинила, акрилонитрила, винилацетата, метакрилата, метилметакрилата. Образование инициаторов реакции - свободных радикалов Начало реакции: образование свободных радикалов, инициирующих начало реакции. +R* + CН2 = C Н2 + CН2 = C Н2; CН2 = C Н2——> R-СН2 –СН2 * ————> R-СН2 –СН2 —СН2 –СН2*————>…; Свободные радикалы могут быть получены несколькими путями: а) с участием инициаторов б) фотолитическим путем в) окислительно-восстановительной реакцией Наиболее распространенными инициаторами являются перекиси органических соединений и азосоединения. Перекиси обычно присутствуют в смесях, которые используют для приготовления полимерных изделий в стоматологии (при пломбировании, протезировании). Под влияние УФ-облучения происходит разры химический связи и образуются свободные радикалы УФ R – O – O– R ———> 2 R – O• (перекись алкила УФ); С6 Н5 – C– O– O– C – С 6Н5 ———> С6 Н5 – C– O• + С 6Н5• + СО2
Механизм радикальной полимеризации может быть представлен на примере 1,4–присоединения. Инициирование Таким образом, в результате актов многократного присоединения растущего радикала к алкадиену происходит рост цепи будущего полимера. Обрыв цепей осуществляется рекомбинацией или диспропорционированием макрорадикалов. Можно видеть, что растущие радикалы обладают двойственной реакционной способностью, что открывает возможность образования 1,4– и 1,2–полимера. Обычно при свободнорадикальной полимеризации доминирующим является 1,4–полимер с примесью 1,2–полимера, причем первый в основном состоит из транс-изомера. В качестве инициаторов этих реакций используют пероксиды или азобисизобутиронитрил. В присутствии металлического натрия осуществляется полимеризация по анион-радикальному механизму: Обозначая можно представить последующий путь полимеризации совокупностью стадий: Полимеризация обычно протекает на поверхности металла, поэтому из-за стерических требований к реакции преимущественно образуется 1,2–полимер. Полимеризация по Циглеру-Натта приводит к образованию стереорегулярного каучука, причем в основном образуются цис-полиалкадиены.
63). Галогенопроизводные углеводородов. Изомерия, номенклатура, получение. Галогенпроизводные углеводородов можно рассматривать как результат замещения однако или более атомов водорода в углеводороде на атомы галогенов. В зависимости от характера связи С-Hal различают следующие типы галогенуглеводородов: 1. Галогенпроизводные со связью С(sp3)-Hal
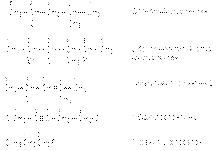
 где R, R/, R// - атом водорода или алкильной радикал.
где R, R/, R// - атом водорода или алкильной радикал.
2. Галогенпроизводные со связью С(sp2)-Hal  3. Галогенпроизводные со связью С(sp1)-Hal R-Cº C-Hal
3. Галогенпроизводные со связью С(sp1)-Hal R-Cº C-Hal
Изомерия. Cтруктурная изомерия. Изомерия положения заместителей; 1-бромбутан - 2-бромбутан. Изомерия углеродного скелета; 1-хлорбутан - 2-метил-1-хлорпропан; Пространственная изомерия Стереоизомерия может проявляться при наличии четырёх разных заместителей у одного атома углерода (энантиомерия) или при наличии разных заместителей при двойной связи, например:транс-1,2-дихлорэтен - цис-1,2-дихлорэтен.Классификация и номенклатура К галогенпроизводным углеводородов со связью С(sp3)-Hal относятся: а) галогеналканы СnH2n+1 Hal, СnH2n+2–x Halx, где х=0....... 2n+2 б) галогенциклоалканы СnH2n-1 Hal, СnH2n–х Halх, где х=0...... 2n в) галогеналкены с атомом галогена в алкильном заместителе при двойной связи 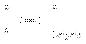 г) галогеналкины с атомом галогена в алкильном заместителе при тройной связи R-Cº C-СnH2n- Hal д) галогенарены с атомом галогена в боковой алкильной цепи ArCnH2n – Hal, ArCnH2n+1–xHalх, где х=0...... 2n+1 В основе названия галогенуглеводорода по номенклатуре IUPAC лежит название самой длинной неразветвленной цепи. Атомы углерода нумеруют таким образом, чтобы меньший номер получил заместитель, который в названии пишется первым, а сами заместители перечисляются в алфавитном порядке. Цепи углеродных атомов в галогенпроизводных алкенов и алкинов нумеруют с того конца, к которому ближе расположена кратная связь Для некоторых простейших галогенпроизводных углеводородов сохраняются названия, в основе которых лежит название углеводородного остатка СH3Cl – метилхлорид, СH3J – метилиодид, С2Н5Вr – этилбромид, С6H5CHCl2 – бензилиденхлорид СH2Cl2 – метиленхлорид, С6H5CH2Cl – бензилхлорид, Для некоторых галогенпроизводных сохраняются тривиальные названия: СHCl3 – хлороформ, СHBr3 –бромоформ, CHJ3 – иодоформ Для названия полностью галогенированных углеводородов используется префикс пер–: С2F6 – перфторэтан C3Cl8 – перхлорпропан Для соединения СCl4 применяется название тетрахлорид углерода и четыреххлористый углерод. Методы получения 1. Заместительное галогенирование. Объектом заместительного галогенирования могут быть алканы. В общем виде реакции заместительного галогенирования алканов выражаются уравнением: RH + Hal2 = RHal + HНal Таким способом можно получить фтор-, бром- и хлоралканы. Подробно об особенностях этих реакций – см. Химические свойства алканов. Аллильное хлорирование алканов можно осуществить при высоких температурах (400-500оС) в паровой фазе
г) галогеналкины с атомом галогена в алкильном заместителе при тройной связи R-Cº C-СnH2n- Hal д) галогенарены с атомом галогена в боковой алкильной цепи ArCnH2n – Hal, ArCnH2n+1–xHalх, где х=0...... 2n+1 В основе названия галогенуглеводорода по номенклатуре IUPAC лежит название самой длинной неразветвленной цепи. Атомы углерода нумеруют таким образом, чтобы меньший номер получил заместитель, который в названии пишется первым, а сами заместители перечисляются в алфавитном порядке. Цепи углеродных атомов в галогенпроизводных алкенов и алкинов нумеруют с того конца, к которому ближе расположена кратная связь Для некоторых простейших галогенпроизводных углеводородов сохраняются названия, в основе которых лежит название углеводородного остатка СH3Cl – метилхлорид, СH3J – метилиодид, С2Н5Вr – этилбромид, С6H5CHCl2 – бензилиденхлорид СH2Cl2 – метиленхлорид, С6H5CH2Cl – бензилхлорид, Для некоторых галогенпроизводных сохраняются тривиальные названия: СHCl3 – хлороформ, СHBr3 –бромоформ, CHJ3 – иодоформ Для названия полностью галогенированных углеводородов используется префикс пер–: С2F6 – перфторэтан C3Cl8 – перхлорпропан Для соединения СCl4 применяется название тетрахлорид углерода и четыреххлористый углерод. Методы получения 1. Заместительное галогенирование. Объектом заместительного галогенирования могут быть алканы. В общем виде реакции заместительного галогенирования алканов выражаются уравнением: RH + Hal2 = RHal + HНal Таким способом можно получить фтор-, бром- и хлоралканы. Подробно об особенностях этих реакций – см. Химические свойства алканов. Аллильное хлорирование алканов можно осуществить при высоких температурах (400-500оС) в паровой фазе 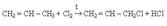 Для аллильного бромирования алканов в качестве реагента используют бромсукцинимид
Для аллильного бромирования алканов в качестве реагента используют бромсукцинимид 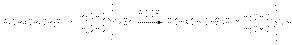 Хлорирование и бромирование боковых цепей алкиларенов можно осуществлять в условиях инициирования свободнорадикальных реакций (химические инициаторы, фотоинициирование), причем наиболее реакционноспособным оказывается a -положения боковой цепи:
Хлорирование и бромирование боковых цепей алкиларенов можно осуществлять в условиях инициирования свободнорадикальных реакций (химические инициаторы, фотоинициирование), причем наиболее реакционноспособным оказывается a -положения боковой цепи: 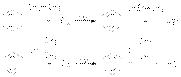
2. Присоединительное галогенирование алкенов и алкинов. Возможности этого метода можно проиллюстрировать следующими реакциями: 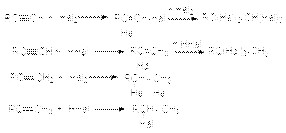 Особенности приведенных реакций подробно обсуждались в разделах, посвященных химическим свойствам алкенов и алкинов. Здесь лишь отметим, что при реализации этих реакций в промышленности с целью форсирования процессов используют катализаторы типа кислот Льюиса (FeCl3, SnCl4, SbCl3 и др.) 3. Реакции замещения гидроксильных групп на галоген в спиртах. К таким реакциям относятся: а) взаимодействие галогеноводородов со спиртами
Особенности приведенных реакций подробно обсуждались в разделах, посвященных химическим свойствам алкенов и алкинов. Здесь лишь отметим, что при реализации этих реакций в промышленности с целью форсирования процессов используют катализаторы типа кислот Льюиса (FeCl3, SnCl4, SbCl3 и др.) 3. Реакции замещения гидроксильных групп на галоген в спиртах. К таким реакциям относятся: а) взаимодействие галогеноводородов со спиртами 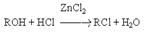 Важным фактором эффективности этих реакций является высокая кислотность среды, обеспечивающая стабилизацию уходящей гидроксильной группы:
Важным фактором эффективности этих реакций является высокая кислотность среды, обеспечивающая стабилизацию уходящей гидроксильной группы: 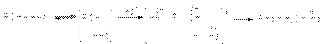 Каталитическое действие хлорида цинка связано с образованием более сильной протонной кислоты за счет донорно-акцепторного взаимодействия:
Каталитическое действие хлорида цинка связано с образованием более сильной протонной кислоты за счет донорно-акцепторного взаимодействия: 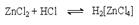 которая в последующем выполняет каталитическую функцию. В связи с важностью поддержания кислотности среды в этих реакциях в качестве гидрогалогенирующих реагентов можно использовать комбинации соль-кислота, например, KBr + H2SO4. Иногда в качестве реагентов используют смеси, продуцирующие in situ необходимый галогеноводород: 2P + 3J2 + 6H2O = 6HJ + 2H3PO3 б) Взаимодействие галогенангидридов кислот (PСl3, PВr3, PСl5, SOCl2, PJ3 и др.) со спиртами: PCl3 + 3ROH = 3RCl + H3PO3; PCl5 + 5ROH = 5RCl + H3PO4 + H2O; SOCl2 + ROH = RCl + SO2 + HCl; SF4 + 2KOH = 2RF + SO2 + 2HF в) Взаимодействие галогенангидридов кислот (PСl5, PBr5, SF4) c альдегидами и кетонами
которая в последующем выполняет каталитическую функцию. В связи с важностью поддержания кислотности среды в этих реакциях в качестве гидрогалогенирующих реагентов можно использовать комбинации соль-кислота, например, KBr + H2SO4. Иногда в качестве реагентов используют смеси, продуцирующие in situ необходимый галогеноводород: 2P + 3J2 + 6H2O = 6HJ + 2H3PO3 б) Взаимодействие галогенангидридов кислот (PСl3, PВr3, PСl5, SOCl2, PJ3 и др.) со спиртами: PCl3 + 3ROH = 3RCl + H3PO3; PCl5 + 5ROH = 5RCl + H3PO4 + H2O; SOCl2 + ROH = RCl + SO2 + HCl; SF4 + 2KOH = 2RF + SO2 + 2HF в) Взаимодействие галогенангидридов кислот (PСl5, PBr5, SF4) c альдегидами и кетонами 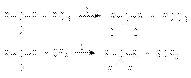 где R’=H, R, Ar. 4. Замещение галогена на галоген. Замещение более легкого галогена в галогенуглеводороде более тяжелым RCl + KBr = RВr + KCl обеспечивается проведением реакции при умеренных температурах и избытке галогенирующего реагента в неполярных апротонных растворителях. Замещение более тяжелого галогена более легким RJ + KCl = RCl + KJ обеспечивается проведением реакции при повышенных температурах и избытке галогенирующего реагента в высокополярной среде в присутствии катализатров - кислот Люиса, например, Сu+, Ag+ и пр. 5. Галогенметилирование аренов
где R’=H, R, Ar. 4. Замещение галогена на галоген. Замещение более легкого галогена в галогенуглеводороде более тяжелым RCl + KBr = RВr + KCl обеспечивается проведением реакции при умеренных температурах и избытке галогенирующего реагента в неполярных апротонных растворителях. Замещение более тяжелого галогена более легким RJ + KCl = RCl + KJ обеспечивается проведением реакции при повышенных температурах и избытке галогенирующего реагента в высокополярной среде в присутствии катализатров - кислот Люиса, например, Сu+, Ag+ и пр. 5. Галогенметилирование аренов 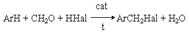
====================================================================
64). Химические свойства галоген производных алканов. Механизмы реакций нуклеофильного замещения. Конкурирующие реакции отщепления. Химическое поведение галогенуглеводородов определяется такими факторами как энергия связи С–Hal, полярность этой связи и ее поляризуемость. Так, относительная слабость связей С–Cl, C–Br и СJ обусловливает их предпочтительное гомолитическое расщепление по сравнению со связями С–С и С–Н. В то же время полярность связей С–Hal и их более высокая поляризуемость по сравнению со связями С–С и С–Н является предпосылкой для их гетеролитического расщепления. Приводимые ниже реакции являются иллюстрацией этих положений. 1. Замещение галогена на водород. Восстановление галогенпроизводных до углеводородов осуществляется водородом в присутствии обычных катализаторов гидрирования (Ni, Pt, Pd) 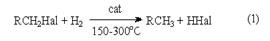 : Реакция имеет гомолитический характер, связанный с предварительной сорбцией реагентов на поверхности катализатора, причем водород претерпевает диссоциативную адсорбцию с вовлечением адсорбированных атомов водорода в реакцию восстановления
: Реакция имеет гомолитический характер, связанный с предварительной сорбцией реагентов на поверхности катализатора, причем водород претерпевает диссоциативную адсорбцию с вовлечением адсорбированных атомов водорода в реакцию восстановления 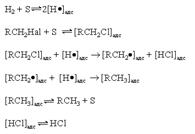 где S – поверхность катализатора. Реакция (1) имеет важное практическое значение как метод переработки галогенорганических отходов в промышленном органическом синтезе. В отличие от другого метода обезвреживания галогенорганических отходов, сжигания, этот метод является ресурсосберегающим и экологически безопасным, так как при его реализации регенерируется углеводородная составляющая исходного сырья и исключается образование высокотоксичных полихлордибензодиоксинов и полихлордибензофуранов. Другой метод замещения галогена на водород – взаимодействие галогенпроизводных углеводородов с иодоводородной кислотой при нагревании
где S – поверхность катализатора. Реакция (1) имеет важное практическое значение как метод переработки галогенорганических отходов в промышленном органическом синтезе. В отличие от другого метода обезвреживания галогенорганических отходов, сжигания, этот метод является ресурсосберегающим и экологически безопасным, так как при его реализации регенерируется углеводородная составляющая исходного сырья и исключается образование высокотоксичных полихлордибензодиоксинов и полихлордибензофуранов. Другой метод замещения галогена на водород – взаимодействие галогенпроизводных углеводородов с иодоводородной кислотой при нагревании 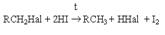 Метод имеет препаративное значение. 2. Взаимодействие с металлами. а) реакция димеризации (синтез Вюрца) 2RHal + Na = R–R + 2NaHal Механизм этой реакции может быть представлен следующей последовательностью стадий:
Метод имеет препаративное значение. 2. Взаимодействие с металлами. а) реакция димеризации (синтез Вюрца) 2RHal + Na = R–R + 2NaHal Механизм этой реакции может быть представлен следующей последовательностью стадий: 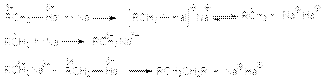 Такой характер взаимодействия является выражением природы реагентов. Натрий как очень активный металл легко отдает свой электрон молекуле галогенуглеводорода, которая выступает в роли слабого электрофила. Электрофильные свойства молекулы R–Hal вытекают из полярности связи С–Hal, обусловливающей дефицит электронов на атоме углерода. Образующийся интермедиат распадается на алкильный радикал и натриевую соль галогена, причем прочная ионная связь в последней обусловливает легкость этого распада. Аналогично можно объяснить энергетическую выгодность последующих реакций механизма. Реакция имеет препаративное значение. б) Взаимодействие с магнием (реакция Гриньяра).
Такой характер взаимодействия является выражением природы реагентов. Натрий как очень активный металл легко отдает свой электрон молекуле галогенуглеводорода, которая выступает в роли слабого электрофила. Электрофильные свойства молекулы R–Hal вытекают из полярности связи С–Hal, обусловливающей дефицит электронов на атоме углерода. Образующийся интермедиат распадается на алкильный радикал и натриевую соль галогена, причем прочная ионная связь в последней обусловливает легкость этого распада. Аналогично можно объяснить энергетическую выгодность последующих реакций механизма. Реакция имеет препаративное значение. б) Взаимодействие с магнием (реакция Гриньяра). 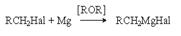 Механизм этой реакции в части ее инициирования подобен механизму реакции Вюрца
Механизм этой реакции в части ее инициирования подобен механизму реакции Вюрца 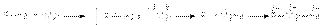 Реактивы Гриньяра имеют важнейшее значение в препаративной органической химии, поскольку открывают возможности получения широкого разнообразия органических соединений на основе взаимодействия этих реактивов с различными реагентами. 3. Реакции нуклеофильного замещения Реакции нуклеофильного замещения – наиболее типичный круг реакций, в которых галогенуглеводороды выступают в качестве субстратов. Результатом этих реакций является замещение галогена на другой атом или группу, которые либо непосредственно выступают в роли нуклеофильного реагента, либо входят в его состав в качестве фрагмента. Наиболее типичными реакциями нуклеофильного замещения галогеналканов и других галогенпроизводных являются: а) реакции гидролиза RHal + H2O = ROH + HHal RHal + NaOH = ROH + NaHal б) реакции образования простых эфиров (реакция Вильямсона) RHal + R`ONa = ROR` + NaHal в) синтез сложных эфиров R1Hal + RCOONa ® RCOOR1 + NaHal г) аммонолиз RHal + 2NH3 = RNH2 + NH4Hal д) синтез нитросоединений и нитритов
Реактивы Гриньяра имеют важнейшее значение в препаративной органической химии, поскольку открывают возможности получения широкого разнообразия органических соединений на основе взаимодействия этих реактивов с различными реагентами. 3. Реакции нуклеофильного замещения Реакции нуклеофильного замещения – наиболее типичный круг реакций, в которых галогенуглеводороды выступают в качестве субстратов. Результатом этих реакций является замещение галогена на другой атом или группу, которые либо непосредственно выступают в роли нуклеофильного реагента, либо входят в его состав в качестве фрагмента. Наиболее типичными реакциями нуклеофильного замещения галогеналканов и других галогенпроизводных являются: а) реакции гидролиза RHal + H2O = ROH + HHal RHal + NaOH = ROH + NaHal б) реакции образования простых эфиров (реакция Вильямсона) RHal + R`ONa = ROR` + NaHal в) синтез сложных эфиров R1Hal + RCOONa ® RCOOR1 + NaHal г) аммонолиз RHal + 2NH3 = RNH2 + NH4Hal д) синтез нитросоединений и нитритов  е) синтез нитрилов и изонитрилов
е) синтез нитрилов и изонитрилов  ж) синтез тиолов и сульфидов R–Hal + NaSH = RSH + NaHal; R–Hal + R1SNa = RSR1 + NaHal; 2R–Hal + Na2S = RSR + 2NaHal з) синтез фосфорорганических соединений
ж) синтез тиолов и сульфидов R–Hal + NaSH = RSH + NaHal; R–Hal + R1SNa = RSR1 + NaHal; 2R–Hal + Na2S = RSR + 2NaHal з) синтез фосфорорганических соединений  (CH3)2PH + CH3CH2Br + NaOH ® (CH3)2PCH2CH3 + NaBr + H2O (диметилэтилфосфин)
(CH3)2PH + CH3CH2Br + NaOH ® (CH3)2PCH2CH3 + NaBr + H2O (диметилэтилфосфин)  и) синтез углеводородов на основе Mg-органических соединений R–MgHal + R’Hal = R–R’ + MgHal2 к) замещение галогена на галоген RX + NaY ® RY + NaY
и) синтез углеводородов на основе Mg-органических соединений R–MgHal + R’Hal = R–R’ + MgHal2 к) замещение галогена на галоген RX + NaY ® RY + NaY
Реакции SN1 Механизм реакции SN1 или реакции мономолекулярного нуклеофильного замещения включает следующие стадии: 1. Ионизация субстрата с образованием карбкатиона (медленная стадия): R−X → R+ + X− 2. Нуклеофильная атака карбкатиона (быстрая стадия): R+ + Y− → R−Y или (если в качестве нуклеофила выступает нейтральная частица): R+ + Y−Z → R−Y+−Z 3. Отщепление катиона (быстрая стадия): R−Y+−Z → R−Y + Z+ Примером реакции SN1 является гидролиз трет-бутилбромида: 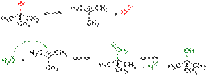 Условный энергетический профиль реакции мономолекулярного нуклеофильного замещения представлен на диаграмме[3]. Скорость реакции SN1 (в упрощённом виде) не зависит от концентрации нуклеофила и прямо пропорциональна концентрации субстрата[4]: Скорость реакции = k × [RX] Так как в процессе реакции образуется карбкатион, его атака (в идеальных условиях без учёта фактора влияния заместителей) нуклеофилом может происходить с обеих сторон, что приводит к рацемизации образующегося продукта. Важно иметь в виду, что SN1 механизм реализуется только в случае относительной устойчивости промежуточного карбкатиона, поэтому по такому пути, обычно, реагируют только третичные ((R)3C-X) и вторичные ((R)2CH-X) алкилпроизводные. Реакции SN2 Механизм реакции SN2 или реакции бимолекулярного нуклеофильного замещения (англ. substitution nucleophilic bimolecular) происходит в одну стадию, без промежуточного образования интермедиата. При этом атака нуклеофила и отщепление уходящей группы происходит одновременно: R−X + Y− → [Y⋯R⋯X]− → R−Y + X− Примером реакции SN2 является гидролиз этилбромида:
Условный энергетический профиль реакции мономолекулярного нуклеофильного замещения представлен на диаграмме[3]. Скорость реакции SN1 (в упрощённом виде) не зависит от концентрации нуклеофила и прямо пропорциональна концентрации субстрата[4]: Скорость реакции = k × [RX] Так как в процессе реакции образуется карбкатион, его атака (в идеальных условиях без учёта фактора влияния заместителей) нуклеофилом может происходить с обеих сторон, что приводит к рацемизации образующегося продукта. Важно иметь в виду, что SN1 механизм реализуется только в случае относительной устойчивости промежуточного карбкатиона, поэтому по такому пути, обычно, реагируют только третичные ((R)3C-X) и вторичные ((R)2CH-X) алкилпроизводные. Реакции SN2 Механизм реакции SN2 или реакции бимолекулярного нуклеофильного замещения (англ. substitution nucleophilic bimolecular) происходит в одну стадию, без промежуточного образования интермедиата. При этом атака нуклеофила и отщепление уходящей группы происходит одновременно: R−X + Y− → [Y⋯R⋯X]− → R−Y + X− Примером реакции SN2 является гидролиз этилбромида: 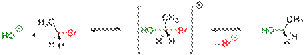 Условный энергетический профиль реакции бимолекулярного нуклеофильного замещения представлен на диаграмме. Скорость реакции SN2 зависит как от концентрации нуклеофила, так и концентрации субстрата: Скорость реакции = k × [RX] × [Y] Так как в процессе реакции атака нуклеофилом может происходить только с одной стороны, результатом реакции является стереохимическая инверсия образующегося продукта. SN2 правило: В реакциях бимолекулярного нуклеофильного замещения атакующий нуклеофил стереохимически инвертирует молекулу, в которой он замещает уходящую группу. Реакции смешанного типа SN1 — SN2 Не для всех реакций можно чётко определить механизм, по которому они протекают, так как чистый SN1 или SN2 являются всего лишь идеальными (предельными) модельными случаями. Следует помнить, что один и тот же субстрат может реагировать с одним и тем же нуклеофилом, в зависимости от условий реакции и растворителя, как по механизму SN1, так и SN2. Например, скорость гидролиза 2-бромпропана описывается с учётом смешанного механизма его протекания[7]: CH3−CHBr−CH3 + HO− → CH3−CHOH−CH3 + Br−; Скорость реакции = k1 × [CH3CHBrCH3] + k2 × [CH3CHBrCH3] × [HO-] Часто смешанный механизм провоцируется применением амбидентных нуклеофилов, то есть нуклеофилов, имеющих не менее двух атомов — доноров электронных пар (например: NO2−, CN−, NCO−, SO32− и пр.) Если в субстрате имеется заместитель, находящийся рядом с атакуемым атомом и несущий свободную электронную пару, он может существенно увеличить скорость реакции нуклеофильного замещения и повлиять на её механизм (сохранение конфигурации). В этом случае говорят об анхимерном содействии соседней группы (например: COO−, COOR, OCOR, O−, OR, NH2, NHR, NR2 и пр.) Примером анхимерного содействия может служить гидролиз 2-бромпропионата:
Условный энергетический профиль реакции бимолекулярного нуклеофильного замещения представлен на диаграмме. Скорость реакции SN2 зависит как от концентрации нуклеофила, так и концентрации субстрата: Скорость реакции = k × [RX] × [Y] Так как в процессе реакции атака нуклеофилом может происходить только с одной стороны, результатом реакции является стереохимическая инверсия образующегося продукта. SN2 правило: В реакциях бимолекулярного нуклеофильного замещения атакующий нуклеофил стереохимически инвертирует молекулу, в которой он замещает уходящую группу. Реакции смешанного типа SN1 — SN2 Не для всех реакций можно чётко определить механизм, по которому они протекают, так как чистый SN1 или SN2 являются всего лишь идеальными (предельными) модельными случаями. Следует помнить, что один и тот же субстрат может реагировать с одним и тем же нуклеофилом, в зависимости от условий реакции и растворителя, как по механизму SN1, так и SN2. Например, скорость гидролиза 2-бромпропана описывается с учётом смешанного механизма его протекания[7]: CH3−CHBr−CH3 + HO− → CH3−CHOH−CH3 + Br−; Скорость реакции = k1 × [CH3CHBrCH3] + k2 × [CH3CHBrCH3] × [HO-] Часто смешанный механизм провоцируется применением амбидентных нуклеофилов, то есть нуклеофилов, имеющих не менее двух атомов — доноров электронных пар (например: NO2−, CN−, NCO−, SO32− и пр.) Если в субстрате имеется заместитель, находящийся рядом с атакуемым атомом и несущий свободную электронную пару, он может существенно увеличить скорость реакции нуклеофильного замещения и повлиять на её механизм (сохранение конфигурации). В этом случае говорят об анхимерном содействии соседней группы (например: COO−, COOR, OCOR, O−, OR, NH2, NHR, NR2 и пр.) Примером анхимерного содействия может служить гидролиз 2-бромпропионата: 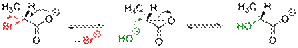 Несмотря на формальный (с точки зрения одностадийности) механизм SN2, образующийся в ходе реакции продукт имеет ту же оптическую конфигурацию, что и исходный. Реакции SNi Механизм реакции SNi или реакции внутримолекулярного нуклеофильного замещения (англ. substitution nucleophilic internal) протекает в несколько стадий по аналогии с механизмом SN1, однако часть уходящей группы при этом атакует субстрат, отщепляясь от оставшейся части. Общая схема реакции:
Несмотря на формальный (с точки зрения одностадийности) механизм SN2, образующийся в ходе реакции продукт имеет ту же оптическую конфигурацию, что и исходный. Реакции SNi Механизм реакции SNi или реакции внутримолекулярного нуклеофильного замещения (англ. substitution nucleophilic internal) протекает в несколько стадий по аналогии с механизмом SN1, однако часть уходящей группы при этом атакует субстрат, отщепляясь от оставшейся части. Общая схема реакции:
1. Ионизация субстрата: 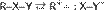 2. Нуклеофильная атака:
2. Нуклеофильная атака: 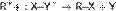 На первом этапе субстрат диссоциирует с образованием т. н. контактной ионной пары. Компоненты такой пары находятся очень близко друг от друга, поэтому атака нуклеофила вынужденно происходит с той же стороны, где до этого находилась уходящая группа. Реакции, протекающие по механизму SNi, крайне редки. Одним из примеров может служить взаимодействие спирта с SOCl2:
На первом этапе субстрат диссоциирует с образованием т. н. контактной ионной пары. Компоненты такой пары находятся очень близко друг от друга, поэтому атака нуклеофила вынужденно происходит с той же стороны, где до этого находилась уходящая группа. Реакции, протекающие по механизму SNi, крайне редки. Одним из примеров может служить взаимодействие спирта с SOCl2: 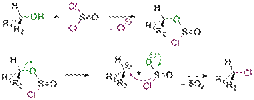 Из схемы видно, что в реакций SNi конфигурация реакционного центра остается неизменной.
Из схемы видно, что в реакций SNi конфигурация реакционного центра остается неизменной.