2015-06-05
2015-06-05 955
955Конкуренция реакций нуклеофильного замещения и отщепления и роль изомеризации Если структура субстрата делает возможным его расщепление, эта реакция всегда конкурирует с замещением. Очевидно, что состав продуктов будет зависеть от соотношения скоростей этих конкурирующих реакций, определяемых различными факторами. При конкуренции SN1– и Е1-реакций 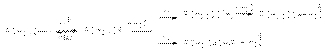 cостав продуктов не зависит от общей скорости и определяется соотношением констант скоростей быстрых стадий k1/k2. На состав продуктов не влияют ни способ образования карбкатиона, ни природа отщепляемой группы Х, что может служить подтверждением SN1– и Е1-механизма. Удлинение и разветвление алкильной группы в исходном соединении приводит к росту доли алкена в продуктах реакции при конкуренции SN1– и Е1-механизмов. Это объясняется большей стабилизацией переходного состояния образования более разветвленного алкена из-за его высокой термодинамической стабильности. переход к более сольватирующей среде способствует увеличению удельного веса продукта замещения, так как переходное состояние быстрой стадии реакции SN1 характеризуется большей степенью локализации заряда (RCH2)3Cd+… Od + H2; чем переходное состояние конкурирующей с ней реакции отщепления
cостав продуктов не зависит от общей скорости и определяется соотношением констант скоростей быстрых стадий k1/k2. На состав продуктов не влияют ни способ образования карбкатиона, ни природа отщепляемой группы Х, что может служить подтверждением SN1– и Е1-механизма. Удлинение и разветвление алкильной группы в исходном соединении приводит к росту доли алкена в продуктах реакции при конкуренции SN1– и Е1-механизмов. Это объясняется большей стабилизацией переходного состояния образования более разветвленного алкена из-за его высокой термодинамической стабильности. переход к более сольватирующей среде способствует увеличению удельного веса продукта замещения, так как переходное состояние быстрой стадии реакции SN1 характеризуется большей степенью локализации заряда (RCH2)3Cd+… Od + H2; чем переходное состояние конкурирующей с ней реакции отщепления 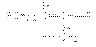 Благодаря этому первое сольватируется сильнее, что приводит к большему понижению энергетического барьера реакции замещения. При конкуренции реакций SN2 и Е2
Благодаря этому первое сольватируется сильнее, что приводит к большему понижению энергетического барьера реакции замещения. При конкуренции реакций SN2 и Е2 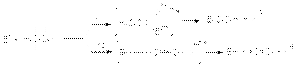
удлинение и разветвление алкильного радикала субстрата приводит к увеличению доли продукта отщепления, как и в случае конкуренции SN1–E1. В основе такого влияния лежат две причины: более высокая термодинамическая стабильность разветвленных алкенов и возрастание стерических препятствий в реакции SN2 с увеличением размера алкильной группы субстрата. В то же время в реакции отщепления Е2 атака основания направлена на периферийный атом водорода и практически не имеет стерических препятствий. Из приведенных соображений следует, что при переводе механизма из SN1– Е1 в SN2– Е2 должен повышаться выход продукта отщепления. В реакции SN2 по сравнению с Е2 переходное состояние характеризуется большей поляризацией отрицательного заряда, поскольку в случае SN2 заряд распределен по трем атомам, а в случае Е2 – по пяти. Поэтому при переходе к более сольватирующему растворителю энергия переходного состояния SN2 будет понижаться в большей степени, чем энергия переходного состояния Е2, что приводит к увеличению доли продукта замещения. Наоборот, при использовании неполярных апротонных растворителей увеличивается выход алкена. Важным фактором регулирования состава продуктов замещения-отщепления является температура. Как правило, энергия активации реакции отщепления (Е2) выше, чем энергия активации замещения (SN2). Поэтому увеличение температуры приводит к увеличению удельного веса продукта отщепления. Понижение температуры приводит к противоположному результату. В случае конкуренции SN1– Е1 температура в меньшей степени влияет на состав продуктов, так как различия в энергиях активации конкурирующих реакций из-за их экзотермичности существенно ниже. Большое значение в конкуренции реакций замещения и отщепления имеет природа нуклеофильного реагента. Успех реакций SN2 определяется его сродством к углеродному атому, имеющему частичный положительный заряд, т.е. нуклеофильностью. При Е2- и Е1св-отщеплении нуклеофил атакует положительно заряженный атом водорода и его активность определяется его основностью. Различие между нуклеофильностью и основностью часто является очень большим и это во многом определяет направление реакции. Для таких сильных оснований, как едкие щелочи и третичные амины наиболее характерны реакции отщепления, что обусловливает их практическое применение для процессов дегидрохлорирование. Наоборот, для относительно сильных нуклеофилов, но слабых оснований (HS–, RS–, ArO–, NH3, Br–, J–) преобладающим направлением является замещение. Этим пользуются при промышленном гидролизе хлорпроизводных в спирты, когда применение едких щелочей дает слишком большой выход алкенов. Замена щелочей на более слабое основание (карбонат натрия, его буферные смеси со щелочью или бикарбонат натрия) позволяет существенно повысить выход спиртов.
65). металлоорганические соединения. Их химические свойства. Использование литий-, натрий-, и магний-органических соединений для синтезов. Металлоорганические соединения (МОС) — органические соединения, в молекулах которых существует связь атома металла с атомом/атомамиуглерода. Щелочные металлы первой подгруппы имеют на внешней электронной оболочке по одному электрону и, следовательно, одновалентны. Металлоорганические соединения построены так, что металл обычно связан поляризованной связью с атомом углерода органического остатка R—Me(где R — алкил или арил): 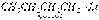
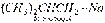
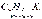 Названия металлоорганических соединений слагаются из названий радикалов и металла. Органические соединения лития. Химические свойства. Вода, спирты, кислоты легко реагируют с литийорганическими соединениями, например:
Названия металлоорганических соединений слагаются из названий радикалов и металла. Органические соединения лития. Химические свойства. Вода, спирты, кислоты легко реагируют с литийорганическими соединениями, например: 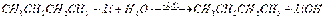 Окисление литийорганических соединений. При регулируемом окислении литийорганические соединения превращаются в спирты:
Окисление литийорганических соединений. При регулируемом окислении литийорганические соединения превращаются в спирты: 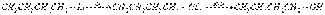 Синтез кислот. Введение литийорганических соединений в избыток двуокиси углерода (их выливают на твердую углекислоту) приводит к литиевым солям карбоновых кислот, которые при действии соляной кислоты превращаются в карбоновые кислоты. Эта реакция широко применяется при исследовании строения литийорганических соединений и для синтеза карбоновых кислот:
Синтез кислот. Введение литийорганических соединений в избыток двуокиси углерода (их выливают на твердую углекислоту) приводит к литиевым солям карбоновых кислот, которые при действии соляной кислоты превращаются в карбоновые кислоты. Эта реакция широко применяется при исследовании строения литийорганических соединений и для синтеза карбоновых кислот: 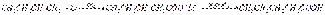 Взаимодействие с непредельными углеводородами — общее свойство для щелочных металлов первой группы. Первоначально считалось, что для успеха реакции двойная связь должна быть сопряжена с непредельной системой или ароматическим кольцом. Однако в 1960 г. К. Циглер показал возможность присоединения литийалкилов к изолированной двойной связи, причем третичные и вторичные литийалкилы реагируют легче первичных. Бутиллитий при нагревании и повышенном давлении присоединяется к этилену с образованием литийалкилов (в которых литий сохраняет высокую реакционную способность):
Взаимодействие с непредельными углеводородами — общее свойство для щелочных металлов первой группы. Первоначально считалось, что для успеха реакции двойная связь должна быть сопряжена с непредельной системой или ароматическим кольцом. Однако в 1960 г. К. Циглер показал возможность присоединения литийалкилов к изолированной двойной связи, причем третичные и вторичные литийалкилы реагируют легче первичных. Бутиллитий при нагревании и повышенном давлении присоединяется к этилену с образованием литийалкилов (в которых литий сохраняет высокую реакционную способность): 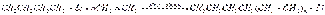
Изопропиллитий реагирует с этиленом уже при -60°С, образуя 1-литий-З-метилбутан: 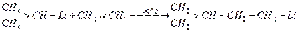 К 1,3-бутадиену литийалкилы присоединяются в положения 1,4 и 1,2. Повышение температуры и давления благоприятствует 1,4- присоединению:
К 1,3-бутадиену литийалкилы присоединяются в положения 1,4 и 1,2. Повышение температуры и давления благоприятствует 1,4- присоединению: 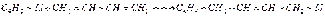
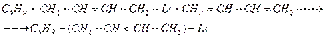 Практическое значение этой реакции заключается в том, что она привела к промышленному методу стереорегулярной полимеризации 1,3-бутадиена в синтетический каучук. Взаимодействие алкиллития с карбонильными соединениями (альдегидами, кетонами), как и в случае натрий-, магний-, цинк-, алюминийорганических соединений, приводит к спиртам. Использование в этой реакции литийорганических соединений оправдано в тех случаях, когда взаимодействие с альдегидами и кетонами более доступных магнийорганических соединений не приводит к цели. Так, диизопропилкетон и изопропиллитий образуют триизопропилкарбинол. Реакция протекает через стадию нестойкого комплекса, который перегруппировывается в литиевый алкоголят, гидролизуемый водой в триизопропил карбинол:
Практическое значение этой реакции заключается в том, что она привела к промышленному методу стереорегулярной полимеризации 1,3-бутадиена в синтетический каучук. Взаимодействие алкиллития с карбонильными соединениями (альдегидами, кетонами), как и в случае натрий-, магний-, цинк-, алюминийорганических соединений, приводит к спиртам. Использование в этой реакции литийорганических соединений оправдано в тех случаях, когда взаимодействие с альдегидами и кетонами более доступных магнийорганических соединений не приводит к цели. Так, диизопропилкетон и изопропиллитий образуют триизопропилкарбинол. Реакция протекает через стадию нестойкого комплекса, который перегруппировывается в литиевый алкоголят, гидролизуемый водой в триизопропил карбинол: 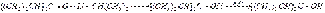 Магнийорганическим синтезом подобный спирт разветвленного строения получить нельзя вследствие восстановления исходного кетона магнийорганическим соединением. Органические соединения натрия Натрийорганические соединения сохраняют ряд общих черт с органическими соединениями лития, однако их специфика заключается: а) в преимущественной роли реакции металлирования при их синтезе, открытой П. П. Шорыгиным (1910 г.) и детально разработанной на примере получения органических соединений натрия; б) в большей реакционной способности, затрудняющей их синтетическое использование. Практическое значение органических соединений натрия связано с инициируемой ими реакцией полимеризации 1,3-бутадиена. Химические свойства. В реакции металлирования углеводороды проявляют свойства слабых кислот. По существу реакция металлирования является реакцией вытеснения слабой кислотой еще более слабой кислоты из ее солей. Порядок вытеснения из натриевых производных углеводородов позволяет составить ряд по возрастающей кислотности:
Магнийорганическим синтезом подобный спирт разветвленного строения получить нельзя вследствие восстановления исходного кетона магнийорганическим соединением. Органические соединения натрия Натрийорганические соединения сохраняют ряд общих черт с органическими соединениями лития, однако их специфика заключается: а) в преимущественной роли реакции металлирования при их синтезе, открытой П. П. Шорыгиным (1910 г.) и детально разработанной на примере получения органических соединений натрия; б) в большей реакционной способности, затрудняющей их синтетическое использование. Практическое значение органических соединений натрия связано с инициируемой ими реакцией полимеризации 1,3-бутадиена. Химические свойства. В реакции металлирования углеводороды проявляют свойства слабых кислот. По существу реакция металлирования является реакцией вытеснения слабой кислотой еще более слабой кислоты из ее солей. Порядок вытеснения из натриевых производных углеводородов позволяет составить ряд по возрастающей кислотности: 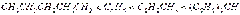 Натрийорганические соединения имеют ионный характер, причем анионом является остаток углеводорода (карбанион), а анионом — металл. Натрийалкилы — сильные основания; так, этилнатрий — сильнейшее из известных оснований. Натрийорганические соединения, как и органические соединения лития, при действии воды, спиртов и кислот разлагаются с замещением натрия водородом:
Натрийорганические соединения имеют ионный характер, причем анионом является остаток углеводорода (карбанион), а анионом — металл. Натрийалкилы — сильные основания; так, этилнатрий — сильнейшее из известных оснований. Натрийорганические соединения, как и органические соединения лития, при действии воды, спиртов и кислот разлагаются с замещением натрия водородом: 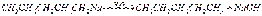 Органические соединения магния Магнийорганические соединения были широко введены в практику органического синтеза В. Гриньяром (1900 г.) и нашли большое применение в органической химии. Образование натнийорганинческих соединений наблюдал за год до этого учитель В. Гриньяра — Ф. Барбье. Получены магнийорганические соединения почти всех классов органических веществ. Синтезы при участии магнийорганических соединений являются одним из важных препаративных методов в органической химии; описано свыше 20 тысяч синтезов при помощи магнийорганических соединений. Способы получения. 1. В среде эфира. Магнийорганические соединения образуются обычно при взаимодействии галогеноалкилов (арилов) с металлическим магнием в среде сухого эфира. Реакция идет при отсутствии влаги. В некоторых случаях реакцию инициируют, добавляя каталитические количества йода или дибромэтана. 2. Безэфирный метод синтеза. Образование магнийорганических соединений из галогеноалкилов и магния катализируется галоидными солями или кислородными соединениями различных металлов (Hg, Al, Sn) и неметаллов (Si, Sb, P) или их алкильными производными. Например, образование магнийорганического соединения в среде ароматических углеводородов (бензола, толуола) инициируется каталитическим количеством тетраэтоксисилана:
Органические соединения магния Магнийорганические соединения были широко введены в практику органического синтеза В. Гриньяром (1900 г.) и нашли большое применение в органической химии. Образование натнийорганинческих соединений наблюдал за год до этого учитель В. Гриньяра — Ф. Барбье. Получены магнийорганические соединения почти всех классов органических веществ. Синтезы при участии магнийорганических соединений являются одним из важных препаративных методов в органической химии; описано свыше 20 тысяч синтезов при помощи магнийорганических соединений. Способы получения. 1. В среде эфира. Магнийорганические соединения образуются обычно при взаимодействии галогеноалкилов (арилов) с металлическим магнием в среде сухого эфира. Реакция идет при отсутствии влаги. В некоторых случаях реакцию инициируют, добавляя каталитические количества йода или дибромэтана. 2. Безэфирный метод синтеза. Образование магнийорганических соединений из галогеноалкилов и магния катализируется галоидными солями или кислородными соединениями различных металлов (Hg, Al, Sn) и неметаллов (Si, Sb, P) или их алкильными производными. Например, образование магнийорганического соединения в среде ароматических углеводородов (бензола, толуола) инициируется каталитическим количеством тетраэтоксисилана: 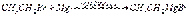 Полученное магнийорганическое соединение далее может быть использовано для синтеза кремнийорганических соединений:
Полученное магнийорганическое соединение далее может быть использовано для синтеза кремнийорганических соединений: 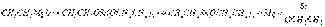 Химические свойства. Магнийорганические соединения не выделяются в свободном виде, обладают высокой реакционной способностью и непосредственно вводятся в дальнейшие реакции. Они вступают в реакции обмена, остаток Mg — Hal замешается водородом, алкильными радикалами, различными металлами и неметаллами. Однако наиболее характерной является реакция присоединения магнийорганических соединений к двойным поляризованным связям. Как раз эти реакции определили выдающуюся роль магнийорганических соединений в органическом синтезе. Основное значение этих реакций заключается в образовании новых углерод-углеродных связей. Взаимодействие с соединениями, содержащими подвижный атом водорода. Вода, спирты, кислоты разлагают магнийорганические соединения с образованием углеводородов:
Химические свойства. Магнийорганические соединения не выделяются в свободном виде, обладают высокой реакционной способностью и непосредственно вводятся в дальнейшие реакции. Они вступают в реакции обмена, остаток Mg — Hal замешается водородом, алкильными радикалами, различными металлами и неметаллами. Однако наиболее характерной является реакция присоединения магнийорганических соединений к двойным поляризованным связям. Как раз эти реакции определили выдающуюся роль магнийорганических соединений в органическом синтезе. Основное значение этих реакций заключается в образовании новых углерод-углеродных связей. Взаимодействие с соединениями, содержащими подвижный атом водорода. Вода, спирты, кислоты разлагают магнийорганические соединения с образованием углеводородов: 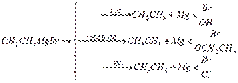 Исходя из этой реакции, Л. А. Чугаевым и Ф. В. Церевитиновым был разработан классический количественный газометрический метод определения подвижного водорода в органических соединениях (метод Чугаева—Церевитинова). Магнийорганические соединения металлируют ацетилен и образуют дибромдимагнийацетилен (Ж. Иоцич):
Исходя из этой реакции, Л. А. Чугаевым и Ф. В. Церевитиновым был разработан классический количественный газометрический метод определения подвижного водорода в органических соединениях (метод Чугаева—Церевитинова). Магнийорганические соединения металлируют ацетилен и образуют дибромдимагнийацетилен (Ж. Иоцич): 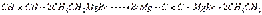 Окисление магнийорганических соединений до спиртов:
Окисление магнийорганических соединений до спиртов: 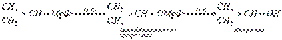 Взаимодействие магнийорганических соединений с альдегидами, кетонами и сложными эфирамизавершается синтезом первичных, вторичных и третичных спиртов. Такой тип превращений магнийорганических соединений имеет наибольшее синтетическое значение преимущественно для получения третичных спиртов. В этом случае магний присоединяется к более электроотрицательному атому кислорода
Взаимодействие магнийорганических соединений с альдегидами, кетонами и сложными эфирамизавершается синтезом первичных, вторичных и третичных спиртов. Такой тип превращений магнийорганических соединений имеет наибольшее синтетическое значение преимущественно для получения третичных спиртов. В этом случае магний присоединяется к более электроотрицательному атому кислорода 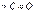 группы, и образовавшиеся алкоголяты гидролизуются водой:
группы, и образовавшиеся алкоголяты гидролизуются водой: 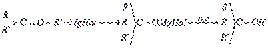 , где:
, где: 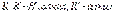 С помощью магнийорганических соединений можно получать также альдегиды, кетоны, кислоты. Магнийорганические соединения используются для синтеза элементоорганических соединений фосфора, кремния, ртути, бора, германия, олова.
С помощью магнийорганических соединений можно получать также альдегиды, кетоны, кислоты. Магнийорганические соединения используются для синтеза элементоорганических соединений фосфора, кремния, ртути, бора, германия, олова.
=========================================================================
66). Спирты. Изомерия, номенклатура. Физические свойства, способы получения. Одноатомные спирты. Классификация, номенклатура, изомерия. Спиртами называются производные углеводородов, представляющие собой продукты замещения атома (атомов) водорода на гидроксильную группу – ОН. В зависимости от того, какое количество атомов водорода замещено, спирты бывают одноатомными и многоатомными. Т.е. число ОН-групп в молекуле спирта характеризует атомность последнего. Наибольшее значение имеют предельные одноатомные спирты, состав которых может быть выражен общей формулой — СnH2n+1ОН или R-OH. Несколько первых членов гомологического ряда спиртов и их названия по радикально-функциональной, заместительной и рациональной номенклатурам приведены ниже: CH3OH - метиловый, метанол, карбинол; C2H5OH - этиловый, этанол, метилкарбинол; н-С3Н7ОН-пропиловый, пропан-1-ол, этилкарбинол; н-С4Н9ОН-бутиловый, бутан-1-ол, пропилкарбинол; н-С5Н11OН-амиловый, пентан-1-ол, бутилкарбинол. По радикально-функциональной номенклатуре название спиртов образуется из названия радикалов и слова «спирт», выражающего функциональное название класса, например, этиловый спирт. Международная IUPAC- номенклатура: к тривиальному названию углеводорода, производным которого является спирт, добавляют окончание –ол (алканолы). Локант указывает номер атома углерода, при котором расположен гидроксил. Главная углеродная цепь выбирается так, чтобы она включала углерод, несущий гидроксильную группу. Начало нумерации цепи так же определяет гидроксил. По рациональной номенклатуре все спирты рассматриваются как производные метанола (СН3ОН), который исторически называется карбинолом, и в котором водородные атомы замещены на один или несколько радикалов. Название спирта составляют из названий радикалов и слова – карбинол (см. табл.14). Таблица 14 Изомерия и номенклатура бутиловых спиртов (С4Н9ОН)
| Cтруктурная формула спирта | Названия спиртов | ||
| радикально- функциональное | международное | рациональное | |
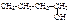 | бутиловый спирт (первичный) | бутан-1-ол | пропилкарбинол |
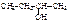 | втор.бутиловыйспирт | бутан-2-ол | метилэтил- карбинол |
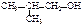 | изобутиловый спирт (первичный) | 2-метил- пропан-1-ол | изопропил-карбинол |
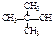 | трет.бутиловый спирт | 2-метил- пропан-2-ол | триметил- карбинол |
Изомерия предельных одноатомных спиртов обусловлена строением углеродного скелета и положением ОН- группы. Метиловый и этиловый спирты не имеют изомеров. В зависимости от положения гидроксильной группы (при первичном, вторичном или третичном С-атоме) спирты могут быть первичными, вторичными или третичными: 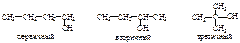 Для бутанола известно 4 изомера (см. таблицу 14). Число изомеров в ряду спиртов быстро растет: С5 – восемь изомеров, С6 – семнадцать, С10 – пятьсот семь. Физические свойства. Газов в гомологическом ряду спиртов нет. Низшие спирты – подвижные жидкости, начиная с С12Н25ОН до С20Н41ОН – маслообразные, а с С21Н43ОН — твердые вещества. Ткип их значительно выше, чем углеводородов с тем же числом С-атомов: ТкипСН3ОН = 65 °С, Ткип С2Н5ОН = 78 °С (ТкипСН4 = –164,5 °С, Ткип С2Н6 = –88,6 °С). Первичные спирты изостроения имеют более низкие температуры кипения, чем нормальные первичные спирты. Более высокие Ткип спиртов обусловлены ассоциацией их молекул друг с другом за счет образования межмолекулярной водородной связи (см. лекция 2). Поэтому метанол – жидкость, а метан – газ. Чтобы разрушить водородные связи, надо затратить энергию, напр. нагреть. Спирты легче воды: их плотности меньше 1,0 (плотность этанола 0,8 г/см3). Метиловый, этиловый и пропиловый спирты смешиваются с водой во всех соотношениях. По мере увеличения и усложнения углеводородных радикалов растворимость спиртов уменьшается. Бутиловый спирт растворяется частично. Высшие спирты в воде не растворяются, т.е. выталкиваются из воды. Многоатомные спирты Двухатомные спирты называются гликолями, трехатомные – глицеринами. По международной заместительной номенклатуре их называются алкандиолами и алкантриолами. Спирты с двумя или тремя гидроксильными группами при одном и том же углеродном атоме обычно в свободном виде не существуют; при попытках их получения они разлагаются, выделяя воду и превращаясь в соединение с карбонильной группой – альдегиды, кетоны или кислоты:
Для бутанола известно 4 изомера (см. таблицу 14). Число изомеров в ряду спиртов быстро растет: С5 – восемь изомеров, С6 – семнадцать, С10 – пятьсот семь. Физические свойства. Газов в гомологическом ряду спиртов нет. Низшие спирты – подвижные жидкости, начиная с С12Н25ОН до С20Н41ОН – маслообразные, а с С21Н43ОН — твердые вещества. Ткип их значительно выше, чем углеводородов с тем же числом С-атомов: ТкипСН3ОН = 65 °С, Ткип С2Н5ОН = 78 °С (ТкипСН4 = –164,5 °С, Ткип С2Н6 = –88,6 °С). Первичные спирты изостроения имеют более низкие температуры кипения, чем нормальные первичные спирты. Более высокие Ткип спиртов обусловлены ассоциацией их молекул друг с другом за счет образования межмолекулярной водородной связи (см. лекция 2). Поэтому метанол – жидкость, а метан – газ. Чтобы разрушить водородные связи, надо затратить энергию, напр. нагреть. Спирты легче воды: их плотности меньше 1,0 (плотность этанола 0,8 г/см3). Метиловый, этиловый и пропиловый спирты смешиваются с водой во всех соотношениях. По мере увеличения и усложнения углеводородных радикалов растворимость спиртов уменьшается. Бутиловый спирт растворяется частично. Высшие спирты в воде не растворяются, т.е. выталкиваются из воды. Многоатомные спирты Двухатомные спирты называются гликолями, трехатомные – глицеринами. По международной заместительной номенклатуре их называются алкандиолами и алкантриолами. Спирты с двумя или тремя гидроксильными группами при одном и том же углеродном атоме обычно в свободном виде не существуют; при попытках их получения они разлагаются, выделяя воду и превращаясь в соединение с карбонильной группой – альдегиды, кетоны или кислоты: 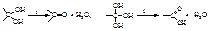 Поэтому первым представителем двухатомных спиртов является производное этана состава С2Н4(ОН)2 с гидроксильными группами при различных углеродных атомах – этан-1,2-диол, или тривиально – этиленгликоль (гликоль). Пропану соответствует уже два двухатомных спирта – пропан-1,2-диол, или пропиленгликоль, и пропан-1,3-диол, или триметиленгликоль:
Поэтому первым представителем двухатомных спиртов является производное этана состава С2Н4(ОН)2 с гидроксильными группами при различных углеродных атомах – этан-1,2-диол, или тривиально – этиленгликоль (гликоль). Пропану соответствует уже два двухатомных спирта – пропан-1,2-диол, или пропиленгликоль, и пропан-1,3-диол, или триметиленгликоль: 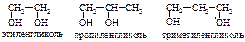
Гликоли, у которых две спиртовые гидроксильные группы расположены в цепи рядом – при соседних атомах углерода, называются a-гликолями (например, этиленгликоль, пропиленгликоль). Гликоли со спиртовыми группами, расположенными через один углеродный атом, называются b-гликолями (триметиленгликоль). Среди двухатомных спиртов этиленгликоль представляет наибольший интерес. В огромных количествах его используют в качестве антифриза для охлаждения цилиндров автомобильных, тракторных и авиационных двигателей; при получении лавсана (полиэфир этиленгликоля с терефталевой кислотой). Этиленгликоль – бесцветная сиропообразная горючая жидкость, не имеющая запаха, сладкая на вкус, ядовита. Смешивается с водой и этанолом в любых соотношениях. Ткип.=197 оС, Тпл.= —13 оС, d420=1,114 г/см3. Дает все реакции, характерные для одноатомных спиртов, причем в них может участвовать одна или обе спиртовые группы. Вследствие наличия двух НО-групп гликоли обладают более кислыми свойствами, чем одноатомные спирты, хотя и не дают кислой реакции на лакмус, не проводят электрического тока. Но в отличие от одноатомных спиртов они растворяют гидроксиды тяжелых металлов. Например, при приливании этиленгликоля к голубому студенистому осадку Cu(OH)2 образуется синий раствор гликолята меди: 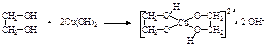 Эта реакция используется для обнаружения a-гликолей. При действии PCl5 обе гидроксидьные группы замещаются на хлор; при действии HCl – замещается только одна НО-группа и образуются так называемые хлоргидрины гликолей:
Эта реакция используется для обнаружения a-гликолей. При действии PCl5 обе гидроксидьные группы замещаются на хлор; при действии HCl – замещается только одна НО-группа и образуются так называемые хлоргидрины гликолей: 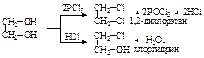 При дегидратации из 2-х молекул этиленгликоля образуется диэтиленгликоль, который может, выделяя внутримолекулярно одну
При дегидратации из 2-х молекул этиленгликоля образуется диэтиленгликоль, который может, выделяя внутримолекулярно одну 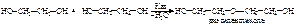

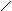
 молекулу воды, превращаться в циклическое соединение с двумя группами простого эфира – диоксан:
молекулу воды, превращаться в циклическое соединение с двумя группами простого эфира – диоксан: 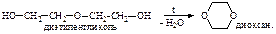 С другой стороны, диэтиленгликоль может реагировать со многими молекулами этиленгликоля, образуя полигликоли – высокомолекулярные соединения, содержащие множество группировок простого эфира:
С другой стороны, диэтиленгликоль может реагировать со многими молекулами этиленгликоля, образуя полигликоли – высокомолекулярные соединения, содержащие множество группировок простого эфира: 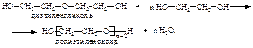 Полигликоли используются в производстве неионогенных синтетических моющих средств, смачивателей, пенообразователей. Этиленгликоль получают щелочным гидролизом 1,2-дихлорэтана, а последний – хлорированием этилена:
Полигликоли используются в производстве неионогенных синтетических моющих средств, смачивателей, пенообразователей. Этиленгликоль получают щелочным гидролизом 1,2-дихлорэтана, а последний – хлорированием этилена: 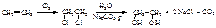 Этиленгликоль может быть получен и прямым окислением этилена водным раствором KMnO4 (реакция Е.Е. Вагнера, 1886), но с небольшим выходом. Глицерин. В природе в свободном виде почти не встречается, но широко распространены его производные – сложные эфиры с высшими карбоновыми кислотами (жиры и масла), которые имеют большое биологическое и практическое значение. Сам глицерин используется в парфюмерии, фармации, в текстильной и пищевой промышленности, для получения нитроглицерина и т.д. Глицерин – бесцветная, вязкая и горючая жидкость, без запаха, сладкая на вкус, не ядовита. Очень гигроскопичен, смешивается с водой и этанолом в любых пропорциях. Ткип. 290 оС (с разложением), d420=1,26 г/см3. Ткип, плотность и вязкость глицерина более высокие, чем у одноатомных спиртов, т.к. он образует больше водородных связей. Это же ведет к более высокой гигроскопичности и растворимости в воде. Глицерин нельзя хранить рядом с сильными окислителями, контакт с которыми приводит к возникновению пожара. Например, взаимодействие с KMnO4, Na2O2, CaOCl2 приводит к самовоспламенению глицерина. Кислотность спиртовых групп в глицерине еще выше, чем у гликоля. В реакциях может участвовать одна, две или три группы. Глицерин также как и этиленгликоль растворяет Cu(OH)2, образуя интенсивно синий раствор глицерата меди. Тем не менее, как и одно– и двухатомные спирты, нейтрален на лакмус. Гидроксильные группы глицерина замещаются на галогены.. При действии водоотнимающих средств или при нагревании глицерин претерпевает дегидратацию. При этом образуется неустойчивый непредельный спирт с гидроксилом при углероде с двойной связью (енол), который изомеризуется в непредельный альдегид акролеин, обладающий резким, раздражающим запахом (как дым пригоревших жиров):
Этиленгликоль может быть получен и прямым окислением этилена водным раствором KMnO4 (реакция Е.Е. Вагнера, 1886), но с небольшим выходом. Глицерин. В природе в свободном виде почти не встречается, но широко распространены его производные – сложные эфиры с высшими карбоновыми кислотами (жиры и масла), которые имеют большое биологическое и практическое значение. Сам глицерин используется в парфюмерии, фармации, в текстильной и пищевой промышленности, для получения нитроглицерина и т.д. Глицерин – бесцветная, вязкая и горючая жидкость, без запаха, сладкая на вкус, не ядовита. Очень гигроскопичен, смешивается с водой и этанолом в любых пропорциях. Ткип. 290 оС (с разложением), d420=1,26 г/см3. Ткип, плотность и вязкость глицерина более высокие, чем у одноатомных спиртов, т.к. он образует больше водородных связей. Это же ведет к более высокой гигроскопичности и растворимости в воде. Глицерин нельзя хранить рядом с сильными окислителями, контакт с которыми приводит к возникновению пожара. Например, взаимодействие с KMnO4, Na2O2, CaOCl2 приводит к самовоспламенению глицерина. Кислотность спиртовых групп в глицерине еще выше, чем у гликоля. В реакциях может участвовать одна, две или три группы. Глицерин также как и этиленгликоль растворяет Cu(OH)2, образуя интенсивно синий раствор глицерата меди. Тем не менее, как и одно– и двухатомные спирты, нейтрален на лакмус. Гидроксильные группы глицерина замещаются на галогены.. При действии водоотнимающих средств или при нагревании глицерин претерпевает дегидратацию. При этом образуется неустойчивый непредельный спирт с гидроксилом при углероде с двойной связью (енол), который изомеризуется в непредельный альдегид акролеин, обладающий резким, раздражающим запахом (как дым пригоревших жиров): 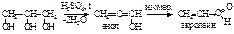 При взаимодействии глицерина с азотной кислотой в присутствии Н2SO4 идет следующая реакция:
При взаимодействии глицерина с азотной кислотой в присутствии Н2SO4 идет следующая реакция: 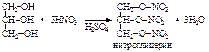 Нитроглицерин – тяжелое масло (d15= 1,601 г/см3), не растворимое в воде, но хорошо растворимое в спирте и других органических растворителях. При охлаждении кристаллизуется (Тпл.=13 оС), очень ядовит. Нитроглицерин – сильное бризантное взрывчатое вещество. Впервые его производство для военных целей организовал Альфред Нобель, заработав себе колоссальное состояние. Проценты от того капитала используются до сих пор в качестве премиального фонда (Нобелевские премии). При ударе и детонации нитроглицерин мгновенно разлагается с выделением огромного количества газов: 4С3Н5(ОNO2)3 ® 12СО2 + 6N2 + О2 + 10Н2О. Для обеспечения безопасности при проведении взрывных работ его используют в виде так называемого динамита – смеси, состоящей из 75 % нитроглицерина и 25 % инфузорной земли (горная порода из кремнистых оболочек диатомовых водорослей). Спиртовый раствор нитроглицерина (1% мас.), не обладающий взрывчатыми свойствами, применяют в качестве сосудорасширяющего средства при лечении болезней сердца. В технике глицерин получают гидролизом (омылением) природных жиров и масел:
Нитроглицерин – тяжелое масло (d15= 1,601 г/см3), не растворимое в воде, но хорошо растворимое в спирте и других органических растворителях. При охлаждении кристаллизуется (Тпл.=13 оС), очень ядовит. Нитроглицерин – сильное бризантное взрывчатое вещество. Впервые его производство для военных целей организовал Альфред Нобель, заработав себе колоссальное состояние. Проценты от того капитала используются до сих пор в качестве премиального фонда (Нобелевские премии). При ударе и детонации нитроглицерин мгновенно разлагается с выделением огромного количества газов: 4С3Н5(ОNO2)3 ® 12СО2 + 6N2 + О2 + 10Н2О. Для обеспечения безопасности при проведении взрывных работ его используют в виде так называемого динамита – смеси, состоящей из 75 % нитроглицерина и 25 % инфузорной земли (горная порода из кремнистых оболочек диатомовых водорослей). Спиртовый раствор нитроглицерина (1% мас.), не обладающий взрывчатыми свойствами, применяют в качестве сосудорасширяющего средства при лечении болезней сердца. В технике глицерин получают гидролизом (омылением) природных жиров и масел: 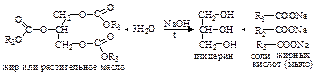 В настоящее время глицерин получают и синтетически из газов крекинга нефти или пропилена. По одному из вариантов синтеза, пропилен хлорируют при высокой температуре (400-500 оС), полученный хлористый аллил путем гидролиза переводят в аллиловый спирт. На спирт действуют перекисью водорода, которая в присутствии катализатора при нагревании присоединяется к аллиловому спирту по двойной связи с образованием глицерина:
В настоящее время глицерин получают и синтетически из газов крекинга нефти или пропилена. По одному из вариантов синтеза, пропилен хлорируют при высокой температуре (400-500 оС), полученный хлористый аллил путем гидролиза переводят в аллиловый спирт. На спирт действуют перекисью водорода, которая в присутствии катализатора при нагревании присоединяется к аллиловому спирту по двойной связи с образованием глицерина: 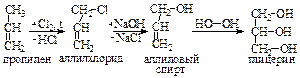
========================================================================
67). Химические свойства спиртов. Кислотно-основные свойства спиртов. Реакции нуклеофильного замещения гидроксо группы. Химические свойства. Кислото-основные свойстыв спиртов. Спирты реагируют со щелочными металлами (Na, K и т.д.) с образованием алкоголятов: 2R—OH + 2Na ® 2R—ONa + H2. Реакция протекает не так бурно, как с водой. Причем с увеличением молярной массы спирта его активность в указанной реакции уменьшается. Первичные спирты значительно активнее в реакциях со щелочными металлами, чем изомерные им вторичные и, особенно, третичные. Спирты в данной реакции проявляют свойства кислот, но они еще более слабые кислоты, чем вода: Ка Н2О=10-16; Ка CH3OH=10-17; КаC2H5OH =10-18. Последнее объясняется электронодонорным влиянием радикала на гидроксильную группу. Как и вода, спирты проявляют и основные свойства, они могут реагировать на холоде с сильными кислотами, образуя соли оксония:  Основные свойства спиртов убывают в противоположном порядке по сравнению с кислотными свойствами, т.е. от третичных к первичным спиртам. Практически же спирты нейтральные вещества: они не показывают ни кислой, ни щелочной реакции на лакмус, не проводят электрического тока. Замещение гидроксильной группы спиртов на галоген возможно при действии галогеноводородов в присутствии серной кислоты или трех- или пятигалоидного фосфора
Основные свойства спиртов убывают в противоположном порядке по сравнению с кислотными свойствами, т.е. от третичных к первичным спиртам. Практически же спирты нейтральные вещества: они не показывают ни кислой, ни щелочной реакции на лакмус, не проводят электрического тока. Замещение гидроксильной группы спиртов на галоген возможно при действии галогеноводородов в присутствии серной кислоты или трех- или пятигалоидного фосфора 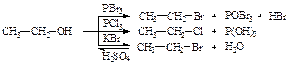 Взаимодействие спиртов с кислотами называют реакцией этерификации. В результате этерификации образуются сложные эфиры:
Взаимодействие спиртов с кислотами называют реакцией этерификации. В результате этерификации образуются сложные эфиры: 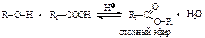 Легче всего этерификации подвергаются первичные спирты, труднее – вторичные и наиболее трудно этерифицируются третичные спирты. Дегидратация спиртов под действием водоотнимающих средств (H2SO4):
Легче всего этерификации подвергаются первичные спирты, труднее – вторичные и наиболее трудно этерифицируются третичные спирты. Дегидратация спиртов под действием водоотнимающих средств (H2SO4):
-внутримолекулярная 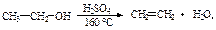 межмолекулярная
межмолекулярная 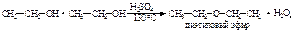 Видно, что результат реакции зависит от условий ее проведения. В первом случае образующаяся вначале при смешивании H2SO4 (избыток) со спиртом алкилсерная кислота при нагревании разлагается, вновь выделяя серную кислоту и этиленовый углеводород. Во втором случае образующаяся вначале алкилсерноя кислота реагирует со второй молекулой спирта с образованием молекулы простого эфира. Окисление спиртов кислородом воздуха возможно при высокой температуре с образованием СО2 и Н2О (процесс горения). Метанол и этанол горят почти бесцветным пламенем, высшие – более ярким коптящим. Это связано с увеличением относительного содержания углерода в молекуле. Кислые растворы KMnO4 и K2Cr2O7 окисляют первичные спирты до альдегидов, а вторичные – до кетонов. При этом малиновый раствор KMnO4 обесцвечивается, а оранжевый раствор K2Cr2O7 становится зеленым [Cr2(SO4)3]. Дальнейшее окисление альдегидов и кетонов приводит к получению карбоновых кислот:
Видно, что результат реакции зависит от условий ее проведения. В первом случае образующаяся вначале при смешивании H2SO4 (избыток) со спиртом алкилсерная кислота при нагревании разлагается, вновь выделяя серную кислоту и этиленовый углеводород. Во втором случае образующаяся вначале алкилсерноя кислота реагирует со второй молекулой спирта с образованием молекулы простого эфира. Окисление спиртов кислородом воздуха возможно при высокой температуре с образованием СО2 и Н2О (процесс горения). Метанол и этанол горят почти бесцветным пламенем, высшие – более ярким коптящим. Это связано с увеличением относительного содержания углерода в молекуле. Кислые растворы KMnO4 и K2Cr2O7 окисляют первичные спирты до альдегидов, а вторичные – до кетонов. При этом малиновый раствор KMnO4 обесцвечивается, а оранжевый раствор K2Cr2O7 становится зеленым [Cr2(SO4)3]. Дальнейшее окисление альдегидов и кетонов приводит к получению карбоновых кислот: 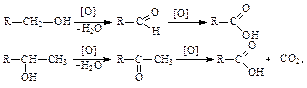 Третичные спирты в мягких условиях устойчивы к действию окислителей, а в жестких – разрушаются, образуя смесь кетонов и карбоновых кислот:
Третичные спирты в мягких условиях устойчивы к действию окислителей, а в жестких – разрушаются, образуя смесь кетонов и карбоновых кислот: 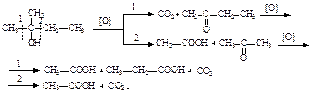 При пропускании паров первичных и вторичных спиртов над поверхностью мелкораздробленных металлов (Cu, Fe) происходит их дегидрирование и образуются карбонильные соединения.
При пропускании паров первичных и вторичных спиртов над поверхностью мелкораздробленных металлов (Cu, Fe) происходит их дегидрирование и образуются карбонильные соединения.
================================================================================
68). Альдегиды и кетоны. Электронное строение карбонильной группы. Методы получения. Представители, применение. Электронное строение альдегидов и кетонов показано на примере муравьиного альдегида: 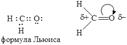 Связь углерод-кислород является одновременно более прочной и более реакционноспособной, чем двойная углерод – углеродная связь. Повышенная реакционная способность связи С=О вызвана различием электроотрицательности углерода и кислорода. В связи с появлением частичного положительного заряда на атоме углерода альдегиды и кетоны проявляют склонность к реакциям с нуклеофильными реагентами. Среди них реакции нуклеофильного присоединения и присоединения - отщепления. Особенности электронного строения служат причиной и других реакций альдегидов и кетонов: протонирование карбонильной группы, СН – кислотность при наличии водородных атомов у α-углеродного атома. Явление СН-кислотности состоит в повышенной подвижности атома водорода в α-положении по отношению к карбонильной группе, вызванной сильным –I-эффектом последней:
Связь углерод-кислород является одновременно более прочной и более реакционноспособной, чем двойная углерод – углеродная связь. Повышенная реакционная способность связи С=О вызвана различием электроотрицательности углерода и кислорода. В связи с появлением частичного положительного заряда на атоме углерода альдегиды и кетоны проявляют склонность к реакциям с нуклеофильными реагентами. Среди них реакции нуклеофильного присоединения и присоединения - отщепления. Особенности электронного строения служат причиной и других реакций альдегидов и кетонов: протонирование карбонильной группы, СН – кислотность при наличии водородных атомов у α-углеродного атома. Явление СН-кислотности состоит в повышенной подвижности атома водорода в α-положении по отношению к карбонильной группе, вызванной сильным –I-эффектом последней: 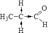 Электронная плотность от атомов водорода смещена на α-углеродный атом, этим вызвана легкость его замещения. Подвижностью атома водорода в α-положении объясняется и явление кето-енольной таутомерии альдегидов и кетонов
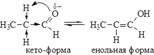
Электронная плотность от атомов водорода смещена на α-углеродный атом, этим вызвана легкость его замещения. Подвижностью атома водорода в α-положении объясняется и явление кето-енольной таутомерии альдегидов и кетонов