2018-02-13
2018-02-13 1735
1735Дикарбоновые кислоты, как и монокарбоновые, делятся на насыщенные, ненасыщенные и ароматические. Номенклатура их также аналогична номенклатуре монокарбоновых кислот:
| Строение кислоты | Название кислоты | ||||
| ИЮПАК | тривиальное | ||||
| НОСО-СООН | Этандиовая | Щавелевая | |||
| НОСО-СН2-СООН | Пропандиовая | Малоновая | |||
| НОСО-СН2-СН2-СООН | Бутандиовая | Янтарная | |||
| НОСО-(СН2)3-СООН | Пентандиовая | Глутаровая | |||
| НОСО-(СН2)4-СООН | Гександиовая | Адипиновая | |||
     Н Н С = С НОСО СООН Н Н С = С НОСО СООН | Цис-бутендиовая | Малеиновая | |||
| Н СООН С = С НОСО Н | Транс-бутендиовая | Фумаровая | |||
НОСО-СºС-СООН | Бутиндиовая | Ацетилендикарбоновая | |||
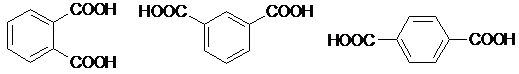
 ИЮПАК 1,2-бензендикар- 1,3-бензендикарбо- 1,4-бензендикарбоновая
ИЮПАК 1,2-бензендикар- 1,3-бензендикарбо- 1,4-бензендикарбоновая
боновая кислота новая кислота кислота
тривиальное фталевая изофталевая терефталевая
Методы синтеза.
17.2.1. Окисление бензена до малеинового ангидрида (лекция № 10).
 |
17.2.2. Окисление нафталина.
Фталевый ангидрид фталевая кислота
17.2.3. Окисление двопервичных гликолей.
17.2.3.1. Окисление этиленгликоля (см. раздел 14.1.3.3).
17.2.3.2. Окисление 1,3-пропиленгликоля.
[O]
 НОСН2-СН2-СН2ОН HOCO-СН2-COOH малоновая кислота
НОСН2-СН2-СН2ОН HOCO-СН2-COOH малоновая кислота
17.2.4. Синтезы на основе малонового эфира (см. раздел 17.3.4).
17.3.1. Кислотность дикарбоновых кислот.
Здесь имеет место двухступенчатая диссоциация:

 НОСO-СООН НОСO-СОО- + Н+ (рКа1)
НОСO-СООН НОСO-СОО- + Н+ (рКа1)
 НОСО-СОО- -ОСО-СОО- + Н+ (рКа2)
НОСО-СОО- -ОСО-СОО- + Н+ (рКа2)
Кислотности сведены в таблице:
| кислота | рКа1 | рКа2 |
| НОСО-СООН | 1,27 | 4,27 |
| НОСО-СН2-СООН | 2,86 | 5,70 |
| НОСО-СН2-СН2-СООН | 4,20 | 5,64 |
| НОСО-СН2-СН2- СН2-СН2-СООН | 4,41 | 5,28 |
| СН3СН2-СООН | 4,87 | - |
Как видим, взаимное влияние карбоксильных групп (-І) приводит к усилению их кислотности (первой степени их диссоциации) в малоновой и особенно в щавелевой кислотах. По мере удаления карбоксильных групп их взаимное влияние снижается, вследствие чего кислотность понижается и постепенно приближается к кислотности монокарбоновых кислот. Анион уменьшает степень диссоциации второго протона (по сравнению с монокарбоновой кислотой), что понятно, если учитывать +I-эффект отрицательно заряженной группы и ее электростатическое влияние напрямую, а не через связи (так называемый эффект поля).
17.3.2. Декарбоксилирование щавелевой и малоновой кислот.
Обе кислоты, особенно малоновая, легко декарбоксилируют при нагревании:
 НОСО-СООН Н-СООН + СО2 (при 150°С)
НОСО-СООН Н-СООН + СО2 (при 150°С)
НОСО-СН2-СООН СН3-СООН + СО2 (плав. с разложением при 136°С).
 |
Механизм разложения малоновой кислоты – образование 6-ти членного (без напряжения) переходного состояния:
Это свойство и используется в синтезах через натрмалоновый эфир. Монокарбоновые кислоты, как уже отмечалось, декарбоксилируются в весьма жестких условиях (без образования 6-членных переходных состояний).
17.3.3. Кето-енольная таутомерия малоновой кислоты и ее эфира и реакции
конденсации с их участием.
Атомы водорода метиленовой группы малоновой кислоты имеют высокую СН кислотность, как и a-водороды в альдегидах и кетонах, особенно в ацетилацетоне СН3-СО-СН2-СО-СН3, где метиленовое звено находится между двумя карбонильными группами, как и в малоновой кислоте.
Чтобы карбоксильные группы свободной малоновой кислоты не мешали реакциям конденсации, малоновую кислоту заменили ее диэтиловым эфиром:
 |
С2Н5О-СО-СН2-СО-ОС2Н5. Катализаторами таких реакций конденсации являются такие сильные основания, как алкоголяты щелочных металлов, например, этилат натрия С2Н5ОNa. Под действием последнего образуется натрмалоновый эфир, по сути своей соль, карбанион которой стабилизирован сопряжением

Однако, енольной формы в малоновом эфире мало (0,08%) по сравнению с 80% для СН3СО-СН2-СОСН3, хотя и больше, чем для ацетона (0,0002%), поскольку анион дестабилизирован сопряжением:
Это сопряжение мешает делокализации отрицательного заряда на углероде, что уменьшает % енола, но практически не мешает реакции конденсации.
17.3.4. Синтез на основе натрмалонового эфира.
17.3.4.1. Синтезы монокарбонових кислот (см. раздел 16.2.2.4).
17.3.4.2. Синтезы дикарбонових кислот.

Пример А:
Пример Б:
 |
17.3.5. Дегидратация.
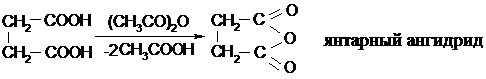 |
Щавелевая и малоновая кислоти воду не отщепляют и ангидриды с неустойчивыми трехчленным и четырехчленным циклами не образуют, и только янтарная кислота легко образует пятичленный циклический ангидрид уже под действием уксусного ангидрида:
 |
Как ангидрид, он реагирует с водой, а с аммиаком образует циклический сукцинимид:
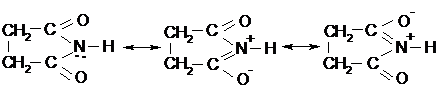 |
В сукцинимиде иминогруппа проявляет слабые кислотные свойства вследствие сопряжения:
 |
Поэтому сукцинимид реагирует со щелочью, образуя натрий сукцинимид, а с бромноватистой кислотой образует бромсукцинимид – мягкий бромирующий агент при аллильном бромировании алкенов (см. раздел 7.3.8 в лекции № 7):
17.3.6. Фумаровая и малеиновая кислоты.
Фумаровая кислота термодинамически более стабильна, чем малеиновая вследствие стерического отталкивания карбоксильных групп в последней:

Фумаровая кислота при нагревании до 270°С превращается в малеиновый ангидрид, который в промышленности главным образом получают окислением бензена. Малеиновый ангидрид в промышленности используется при производстве синтетических смол и полимеров, а также в органическом синтезе, например, он эффективный диенофил в реакции Дильса-Альдера.
17.3.7. Фталевые кислоты.
 |
Соответствующие фталевые кислоты можно получить окислением ксиленов, например, п-ксилена:
 |
Из всех фталевых кислот (их три) только фталевая (о-изомер) способна образовывать ангидрид, который, как все ангидриды, легко гидролизуется, а также реагирует с аммиаком с образованием фталимида:
Фталимид, как и сукцинимид, обладает слабокислым характером и реагирует со щелочами, образуя фталимиды металлов, используемые в качестве промежуточных продуктов в дальнейших синтезах:

Фталимид калия использован в реакции Габриэля – методе получения первичных аминов, свободных от вторичных и третичных.
Практическое применение нашли эфиры фталевой кислоты, получаемые из фталевого ангидрида, например:

Такие эфиры – жидкости с высокой температурой кипения, используются как растворители и пластификаторы полимеров (поливинилхлорида, целлюлозы).
Фталевый ангидрид применяется для получения полиэфирных смол типа глифталевых (из гликолей) или пентафталевых (из пентаэритрита С(СН2ОН)4), которые являются основой эмалей и красок. Пример – глифталевая смола, получаемая из глицерина и фталевого ангидрида:
 |

Терефталевая кислота в виде диметилового эфира (легче очищается в промышленных условиях) используется для синтеза полиэфирного волокна лавсана реакцией переэтерификации:
Лекция № 18.
АЗОТСОДЕРЖАЩИЕ СОЕДИНЕНИЯ.
Здесь несколько классов соединений:
R-NO2; R-NH2; R-CO-NH2; R-N=N-R; R-N+ºN×X-; R-CºN,
нитросо- амины амиды азосоедине- диазосоеди- нитрилы
единения ния нения
гидроксамовые кислоты R-CO-NH-OH.
Нитросоединения.
18.1.1. Классификация и номенклатура нитросоединений.
 |
Общая формула нитросоединений - R-NO2, они делятся в основном на два типа: алифатические и ароматические. Алифаимческие соединения в зависимости от углерода, с которым связана нитрогруппа, делятся на первичные, вторичные и третичные:
Принципы номенклатуры уже освещались в лекции № 3.
18.1.2. Методы получения.
Из всех методов получения уместно упомянуть только реакции нитрования алканов (реакция Коновалова) и аренов (лекции № 10 и 11).
18.1.3. Строение нитрогруппы. Полярность нитросоединений.
 |
Нитрогруппа имеет семиполярную связь, ее строение можно описать двумя резонансными структурами:
Нитросоединения сильно полярны, ибо содержат сильно полярную электроноакцепторную нитрогруппу.
18.1.4. Свойства нитросоединений.
18.1.4.1. Реакции восстановления.
Используются различные восстановители: H2/Pt, HCl + Zn, HCl + Fe, и т.д. Зинин впервые восстановил нитробензол сероводородом:
50oC
 C6H5NO2 + 3H2S C6H5NH2 + 2H2O + 3S.
C6H5NO2 + 3H2S C6H5NH2 + 2H2O + 3S.
При восстановлении в кислой и щелочной средах образуется ряд разных промежуточных продуктов, но конечным продуктом восстановления всегда является амин. Студентам, интересующимся проблемами синтеза промежуточных продуктов восстановления, рекомендуется смотреть учебники и соответствующую литературу.
18.1.4.2. Реакции алифатических нитросоединений со щелочами.
Гиперконьюгация С-Н связей с нитрогруппой обуславливает подвижность водорода (кислотность С-Н связей) в a-положении к нитрогруппе (и только в щелочной среде), что позволяет первичным и вторичным нитросоединениям реагировать со щелочами с образованием солей нитроновых кислот:

Первичные и вторичные нитросоединения называют псевдокислотами, ибо они не диссоциируют на анионы и протон, а отщепляют протон только под действием НО-. Третичные нитросоединения со щелочами не реагируют, ибо не имеют кислого водорода в a-положении к нитрогруппе.
18.1.4.3. Гидролиз солей нитроновых кислот.

Сами нитроновые кислоты в свободном состоянии не существуют. Поэтому при действии разбавленных сильных кислот из первичных нитроновых солей образуются альдегиды, из вторичных – кетоны:
18.1.4.4. Реакции алифатических нитросоединений с азотистой кислотой.
Первичные и вторичные нитросоединения реагируют с азотистой кислотой по разному. В случае первичных соединений образуется соль красного цвета:

В случае вторичных соединений образуется псевдонитрол синего цвета:

Третичные нитросоединения с азотистой кислотой не реагируют.
Первичные и вторичные нитросоединения вступают в реакции конденсации с альдегидами и кетонами в качестве метиленовой компоненты.
18.1.4.5. Реакции первичных алифатических нитросоединений с
концентрированной Н2SO4.

В качестве примера такой реакции можно привести процесс превращения нитроэтана в гидроксиламин и уксусную кислоту в концентрированной Н2SO4 (80 – 90 %) по схеме:
Этим способом в промышленности получают гидроксиламин из нитрометана.
18.1.4.6. Реакции нуклеофильного замещения с участием
нитропроизводных бензола.

Как известно, нитро группа уменьшает способность бензольного кольца к реакциям электрофильного замещения, но в то же время увеличивают его активность к реакциям нуклеофильного замещения. Легко эти реакции идут для тринитропроизводных. Пример:
 |
Эти реакции, как и в случае электрофильного замещения, происходят с образованием s-комплексов, которые в этом случае называют комплексами Мейзенгеймера:
Возможно замещение и атома водорода на нуклеофил, только в этом случае водород должен отщепляться в виде нуклеофила, т.е. Н:- (гидрид-анион), для этого в смеси должен присутствовать окислитель, например, гексацианоферат калия К3Fe(CN)6 (красная кровяная соль):
Пикриновая кислота (2,4,6-тринитрофенол, техническое название мелинит, шимоза) – бризантное (дробящее) взрывчатое вещество. В виду опасности при производстве вытеснена веществами типа тола (тринитротолуола).
 |
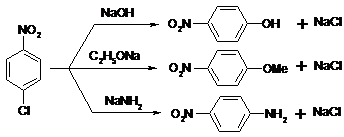 |
Важную роль в органическом синтезе играют реакции нуклеофильного замещения галогена в тех соединениях, которые содержат по крайней мере одну нитрогруппу в о- или п-положениях. Пример:
 |
Увеличение количества нитрогрупп содействует нуклеофильному замещению, например, пикрилхлорид медленно гидролизуется водой уже при комнатной температуре:
Амины.
Амины – соединения общей формулы R-NH2.
18.2.1. Классификация и номенклатура аминов

Амины можно разделить на алифатические (жирные), ароматические и жирноароматические (алкилароматические). Их все можно рассматривать как производные аммиака NH3, в котором атомы водорода замещены на радикалы:
Первичный вторичный третичный четвертичная аммониевая
амин, амин, амин, соль,
метиламин диметиламин триметиламин тетраметиламмоний хлорид.
 |
Приведенные названия образованы по радикально-функциональной номенклатуре. Пример названий амина с разными радикалами:
название по радикально-функциональной номенклатуре – диметилпропиламин, по ИЮПАК – 1-(N,N-диметиламино)пропан.
Названия ароматических аминов:
Амин тривиальное радикально- номенклатура по ИЮПАК

назване функциональ-
ное название
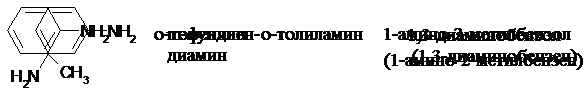 |
18.2.2. Методы получения аминов
Из методов получения уже рассматривались реакции восстановления нитропроизводных (раздел 18. 1.4.1) и синтез Габриеля чистых первичных аминов (раздел 17.3.7). Ниже представлены и другие методы.
 |
18.2.2.1. Алкилирование аммиака по Гофману.
Алкилирование по Гофману – реакция исчерпывающего алкилирования:
Недостаток метода – образуется смесь аминов (в отличие от синтеза Габриеля). Чтобы получить только первичный амин, надо брать большой избыток аммиака.
 |
18.2.2.2. Перегруппировка по Гофману (получение из амидов).
18.2.2.3. Перегруппировка по Бекману.
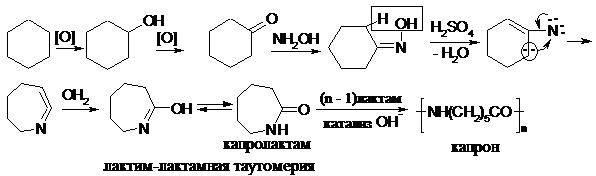
Наблюдается для оксима циклогексанона и используется для получения капролактама (циклического амида), мономера для синтеза полиамида капрона (волокно для чулок):
18.2.3. Физические свойства аминов.

Температуры кипения и плавления первичных аминов всегда выше, чем вторичных, и тем более чем третичных аминов с приблизительно одинаковым молекулярным весом. Это объясняется наличием водородных связей А в первичных, Б во вторичных и отсутствием всяких водородных связей в третичных
аминах. Однако, указанные водородные связи слабее, чем связи ОН× × ×О с участием гидроксила.
18.2.4. Химические свойства аминов.
Химические свойства аминов определяются аминогруппой, атом азота которой имеет неподеленную пару электронов. Поэтому для них в первую очередь характерны свойства оснований и нуклеофильная реакционная способность.
18.2.4.1. Основные свойства аминов.
 |
Это сильные основания, их водные растворы имеют щелочную реакцию:
При взаимодействии аминов с кислотами образуются аммонийные соли:

Алкилы вследствие +І эффекта усиливают основные свойства аминов, поэтому в неводных растворителях амины по основности располагаются в ряд:
Но в водных растворах основность третичных аминов ниже, чем вторичных по двумь причинам:
алкильные заместители создают стерические препятствия для сольватации аминов молекулами воды, которая усиливает основность;
третичные амины не образуют характерные для всех других аминов водородные связи типа N-H× × × OH2, которые также усиливают основность.
 |
Анилин менее основен, чем аммиак, вследствие сопряжения:
Поэтому по основности ароматические амины располагаются в ряд:
NH3 > C6H5-NHCH3 > C6H5-NH2 > C6H5-NH-C6H5 > (C6H5)3N
18.2.4.2. Реакции алкилирования и ацилирования аминов.

Амины легко алкилируются. В качестве примера уже была приведена реакция исчерпывающего алкилирования по Гофману. Реакции ацилирования идут еще легче (студентам задание: объясните, почему?):
18.2.4.3. Реакции аминов с азотистой кислотой.
 |
Очень неустойчивое соединение азотистую кислоту получают непосредственно в реакционной среде во время реакции из соли:
Реакция со вторичным амином останавливается на образовании нитрозамина, третичные амины с азотистой кислотой не реагируют.
18.2.4.4. Образование оснований Шиффа.
Ароматические амины с ароматическими альдегидами образуют основания Шиффа (азометины):

Эти соединения легко гидролизуются в кислой среде:
 |
Поэтому реакцию образования азометинов используют для защиты аминоггруппы в органическом синтезе.
18.2.4.5. Расщепление четвертичных аммониевых солей.
 |
Тетраметильные аммониевые основания не расшепляются. Расщепление начинается с этильных радикалов. Расщеплению подвергаются четвертичные гидроксиды аммония, например:
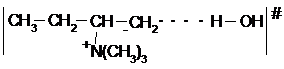 |
Отщепление происходит против правила Зайцева (по правилу Гофмана – водород отщепляется от наиболее гидрогенизированного атома углерода). Хотя здесь все еще остается Е2 механизм, вследствие очень низкой способности такого сильного основания как триметиламин к отщеплению переходное состояние реакции по своему строению близко к карбаниону, так как отщепление протона сильно опережает отщепление триметиламина:
 |
При этом здесь отщепляется наиболее кислый водород группы СН3:
поскольку +І эффект группы СН3 уменьшает положительный заряд на углероде в положении 3 и не достает (ибо затухает) до углерода в положении 1.
Причины отщепления по правилу Зайцева детально обсуждаются в разделе 12.5.4 лекции № 12.
Лекция № 19.
СВОЙСТВА АРОМАТИЧЕСКИХ АМИНОВ. ДИАМИНЫ. АРОМАТИЧЕСКИЕ И АЛИФАТИЧЕСКИЕ ДИАЗОСОЕДИНЕНИЯ.