 2014-02-09
2014-02-09 1057
1057Эквивалентная электрическая проводимость представляет собой проводимость такого объема раствора, который помещен между двумя электродами на расстоянии 1м и содержащего 1 г-экв. вещества.
Если между электродами поместить Vм3 раствора, содержащего 1 г-экв вещества, то между удельной и эквивалентной электропроводностью установится связь:
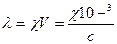
где с – моль/л, 103/с –м3/моль, разведение. Разведение –показывает объем раствора, в котором разведена одна молярная масса эквивалента.
После подстановки значения χ получим, что эквивалентная электрическая проводимость равна сумме подвижностей ионов:
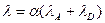
Эквивалентная электропроводность имеет размерность См*м2/моль.
Зависимость эквив. электропроводности слабых и сильных электролитов от концетнрации одинакова. С увеличением разбавления электролита она растет и достигает максимума в области предельного разбавления и достигает предельного значения λ∞.
Предельная эквивалентная электрическая проводимость - λ∞ это электрическая проводимость гипотетического разбавленного раствора, характеризующегося полной диссоциацией электролита и отсутствием сил электростатического взаимодействия. между ионами.

 Согласно χ=
Согласно χ=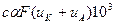 и
и
эквивалентная электрическая проводимость бесконечно разбавленного раствора (степень диссоциации α=1) выражается уравнением:
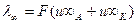
но Fu∞ =λ ∞ -, поэтому:
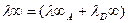
Это соотношение называют законом независимого движения ионов:
В бесконечно разбавленном растворе ионы движутся независимо один от другого.
Этот закон был установлен Ф. Кольраушем. Предельная подвижность ионов является специфической величиной, т.е. зависит от природы иона, природы растворителя и температуры и не зависит от природы другого иона а растворе электролита.
Если разделить на то можно получить выражение для степени диссоциации слабого электролита:
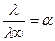
Пользуясь известными значениями предельных подвижностей ионов можно вычислить эквивалентную электрическую проводимость электролитов при бесконечном разведении.
Самыми большими (аномальными) значениями предельных подвижностей обладают ионы гидроксония и гидроксила. Их предельные подвижности находятся в пределах 349,8 и 199,2 а подвижности других ионов находятся в пределах 40-80 См*м2,моль. Это объясняется тем, что движение этих ионов происходит в растворе не только за счет миграции, но и путем передачи иона водорода от одной молекулы воды к другой по эстафетому механизму.
Исчезновение или появление аномально высокоподвижных ионов гидроксония и гидроксила вызывает резкие скачки удельной электропроводности, на этом основан метод кондуктометрического титрования.
Электродные процессы, гальванические элементы. Возникновение электродного потенциала на границе раздела двух фаз.
Уравнение Нернста, индикаторные электроды, электроды сравнения.
Термодинамика гальванического элемента. Электрометрическое определение рН растворов.
Электродом называют электропроводящую фазу (металл ли полупроводник), контактирующую с ионным проводником (электролитом).
Электродные процессы – это о-в реакции, протекающие на электродах.
Электропроводящая фаза вместе с раствором или расплавом образует полуэлемент.
Гальваническим элементом называется устройство, состоящее как минимум из двух полуэлементов, в котором энергия химической реакции превращается в электрическую.
Гальванический элемент характеризуется электродными потенциалами и разностью потенциалов ЭДС.
Современные представления возникновения электродного потенциала дает сольватационная теория, объединяющая результаты исследований В. Нернста, Н.А. Изгарышева, и У. Герни.
Главными процессами в этой цепи являются:
1. Ионизация электродного металла с появлением в нем ионов и свободных электронов. Ионизация электродного металла характеризуется энергией разрушения кристаллической решетки, которая равна работе выхода иона металла из кристаллической решетки.
2. Взаимодействие растворителя с ионами металла, находящимися в кристаллической решетке. Сольватация характеризуется энергией сольватации.
3. Соотношение этих энергий определяет направление суммарной реакции, если энергия сольватации > энергии разрушения кристаллической решетки, то ионы металла будут переходить в раствор, так как все процессы идут в сторону уменьшения энергии. В металле создается избыток электронов, поэтому он заряжается отрицательно. и наоборот, если энергия сольватации будет, работы выхода электрона, то будет происходить переход ионов металла из раствора в металл, при этом они будут терять сольватную оболочку. Металл будет заряжаться положительно, а раствор – отрицательно.
4. Электростатическое взаимодействие между металлом и ионами раствора препятствует беспредельному переходу ионов в одном направлении. В итоге, когда обе энергии будут равны, в системе металл –раствор устанавливается подвижное равновесие. На границе двух фаз формируется двойной электрический слой, которому соответствует определенное значение электрического потенциала.
Поскольку условие равновесия системы является равенство электрохимических потенциалов каждого вида частиц в контактирующих фазах, то можно записать:
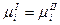 то и
то и 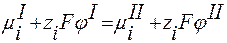
Отсюда скачок потенциала выразится уравнением:
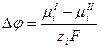
Химический потенциал иона в металле 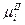 можно считать постоянным, а химический потенциал иона в растворе выражается уравнением:
можно считать постоянным, а химический потенциал иона в растворе выражается уравнением:
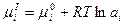
Тогда 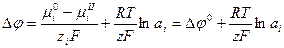
где 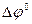 - стандартный потенциал электрода при а =1, а
- стандартный потенциал электрода при а =1, а 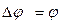
Выражение для потенциала электрода принимает вид:
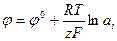
В этом уравнении 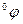 - стандартный потенциал, т.е. потенциал, возникающий при стандартных условиях, т.е. когда а =1.
- стандартный потенциал, т.е. потенциал, возникающий при стандартных условиях, т.е. когда а =1.
Разность потенциалов между точками внутри фаз (гальвани-потенциал) определяется при вычитании одного уравнения из другого:
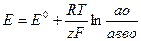
Это уравнение носит название уравнения Нернста. Для металлических электродов, у которых восстановленной формой является металл, знаменатель отбрасывается, так как а металла=1.
6. индикаторные электроды, электроды сравнения.
7. Термодинамика гальванического элемента. Электрометрическое определение рН растворов.
Классификация электродов (самост-но, Стромберг, Киреев)
Термодинамика гальванического элемента.
В гальванических элементах химические реакции на электродах протекают тем медленее, чем большим соротивлением обладает внешняя электрическая цепь (выводы, вольтметр). Принципиально можно замкнуть цепь проводноком бесконечно большого сопротивления, и реакция будет идти бесконечно медленно, так что в каждый момент будет существовать равновесие между электродами и растворами.
Такое течение реакции является обратимым. В случае термодинамически обратимого процесса получается максимальная электрическая работа.
Она равна ЭДС элемента Е, умноженной на переносимый заряд. Если во время реакции произойдет восстановление или окисление n ионов, то по закону М.Фарадея, перенесенный заряд равенnF.
Гальванический элемент работает самопроизвольно, поэтому ΔG < 0, и ΔG=nFЕ, поэтому:

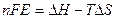 ,
,
в соответствии с тем, что при p=const
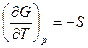
имеем: 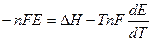 откуда
откуда 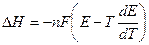
производную 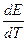 называют температурным коэффициентом ЭДС.
называют температурным коэффициентом ЭДС.
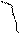




 Зная его и измеряя ЭДС, можно определить тепловой эффект реакции.
Зная его и измеряя ЭДС, можно определить тепловой эффект реакции.
КИНЕТИКА
Принципиальную возможность той ли иной реакции предсказывает химическая термодинамика (ΔG < 0).
Однако не всегда термодинамическивозможные реакции осуществляются в действительности. Например, все органические вещества, согласно принципам термодинамики, должны были бы достаточно быстро окислиться в углекислоту и воду молекулярным кислородом воздуха, так как этот процесс сопровождался бы значительным уменьшением энергии Гиббса.
Существование растений, животных, залежей нефти, ископаемых, угля обязано тем, что реакция окисления протекает в действительности исключительно медленно.
Химическая кинетика устанавливает законы, определяющие скорость химических процессов, и выясняет роль различных факторов, влияющих на скорость и механизм реакций.
Химическая кинетика состоит из двух разделов: формальная кинетика – дает математическое описание скорости реакции без учета механизма самой реакции, и 2) молекулярная кинетика – учение о механизме химического взаимодействия.
Согласно закону действия масс, скорость химической реакции определяется выражением:
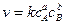 носит название основного постулата химической кинетики.
носит название основного постулата химической кинетики.
Физический смысл константы:
это есть скорость химической неакции при концентрациях реагирующих веществ, равных единице.
Константа, как и скорость, зависит от природы веществ, температуры, наличия катализаторов, но не зависит от концентрации. По известным величинам k сравнивают скорости различных реакций.
Молекулярность и порядок реакции
Молекулярность определяется число молекул, одновременно участвующих в элементарном акте химической реакции. Реакции могут быть одно- двух- или трехмолекулярные.
Порядок реакции определяется сумой показателей степени при концентрациях, входящих в кинетическое уравнение скорости химической реакции
Реакции могут быть (v= первого kc). второго (v=kc2) и тертьего порядка(v=kc3)
Так же может быть нулевой порядок (скорость подвода вещества больше, чем скорость его расходования) и дробный порядок – для сложных реакций, проходящих ряд промежуточных стадий.
Причины несовпадения молекулярности и порядка
1) постоянство концентрации одного из участников реакции. Например, в реакции омыления эфира вода практически имеет постоянную концентрацию:
С2Н5СООСН2 + Н2О = С2Н5ОН +СН3СООН
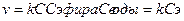
реакция бимолекулярна, но имеет первый порядок.
2)ступенчатый характер реакции:
например, тримолекулярная реакция хлорирования оксида азота
2NO +Cl2 = 2NOCl
состоит итз двух стадий: NO + Cl2 = NOCl2 и NOCl2+NO =2NOCl
Реакции обычно характеризуются кинетическим уравнением, которое позволяет рассчитать константу скорости в любой момент времени от ее начала, и периодом полупревращения, который определяет момент уменьшения первоначальной концентрации реагирующих веществ вдвое.
Каждый порядок реакции имеет свое кинетическое уравнение, позволяющее определить константу скорости:
1) 1-ый порядок: 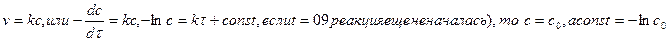 тогда
тогда 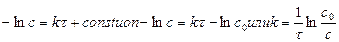
2) аналогично, для 2-го порядка:
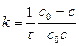
2) для 3-го порядка:
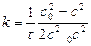
Методы определения порядка реакции
1) метод подстановки. Заключается в экспериментальном определении концентрации вещества в различные момента реакции (титрованием, поляриметрией). По полученным данным производят расчет констант скоростей, подставляя их в уравнения 1-го, 2-го, 3-го порядков.
Выясняют, по какому уравнению расчет дает постоянную величину (в пределах ошибки) с небольшими отклонениями. Этому порядку и подчиняется исследуемая реакция.
2) Графический метод. Экспериментально измерив концентрации вещества в различные промежутки времени, строят графики зависимости функции концентрации (ln C, 1/с, 1/с2) реакция будет того порядка, где график даст прямую линию.
3) По периоду полупревращения. для реакций первого порядка время полупревращения не зависит от начальной концентрации вещества, для 2-го порядка – фяч, дл 3-го – обратно пропорционально квадрату начальной концентрации.
4) метод Вант-Гоффа. v=kcn
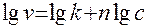
Строят график зависимости логарифма скорости от логарифма концентрации, определяют наклон полученной прямой, он будет представлять собой порядок реакции по тому веществу, концентрация которого измерялась. Отрезок на оси ординат равен логарифму константы.
Зависимость скорости реакции от темпераатуры
Правило Вант-Гоффа: ориентировочно повышение температуры на каждые 10 увеличивает константу скорости в 2-4 раза.
Основан метод искусственного "старения" лекарственных препаратов для определения их срока годности.
Более точную зависимость константы скорости от температуры дает уравнение Аррениуса:
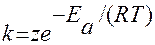 или lnk=B-А/Т по графику lg от 1/Т находят В и А:
или lnk=B-А/Т по графику lg от 1/Т находят В и А:
lnk=lnz - Ea/RT
 |
Энергия активации – избыток энергии по сравнению со средней энергией реагирующих веществ, который необходим для того, чтобы соударения были результативными. (Теория активных соударений).
(Теория активных соударений).
Катализом называют явление изменения скорости реакции в присутствии катализаторов.
Гомогенный – если катализатор и вещества представляют собой одну фазу.
Гетерогенный – если протекают на границе раздела фаз.

