 2015-02-27
2015-02-27 1744
1744Запишем термохимическое уравнение реакции сгорания:
C3H8 + 5O2 = 3CO2 + 4H2O(г); ∆ Hхр
Применяя к исходному уравнению следствие из закона Гесса, получаем:
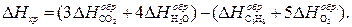
Подставим табличные значения теплот образования, учитывая, что теплоты образования простых веществ приняты равными нулю:
∆ Hхр = 3(–393,51) + 4(–241,83) – (–103,92) = –2043,93 кДж/моль.
Найденное количество тепла выделяется при сгорании одного моля пропана. Напомним, что 1 моль газа при нормальных условиях занимает объем VM = 22,4 л/моль. По условию задачи сгорает 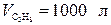 , т. е.:
, т. е.:
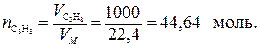
Количество тепла, выделяемого при сгорании 1 м3 пропана, равно:
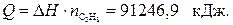
Надо подчеркнуть, что следствие из закона Гесса широко используется также для расчета изменения энтропии и изобарных потенциалов при химических реакциях.
Таблица 2.1
Термодинамические функции D Нобр (кДж/моль) и S 0 (Дж/ моль∙К)
некоторых веществ при стандартных условиях
| Вещество | ∆ Hобр | S 0 | Вещество | ∆ Hобр | S 0 | Вещество | S 0 |
| Al2O3 | –1675 | Fe(OH)2 | –562 | Ag | |||
| Al(OH)3 | –1294 | FeS2 | –177 | Al | |||
| AlN | –318 | HСl(г) | –92 | Ba | |||
| Ag2O | –31 | HNO3(ж) | –174 | C | |||
| AlCl3 | –704 | H2O (ж) | –286 | Ca | |||
| BaO(т) | –554 | H2O (г) | –242 | Cl2 | |||
| CaCO3 | –1206 | H2S (г) | –21 | Cr | |||
| CaO(т) | –635 | H2SO4 (ж) | –814 | Cu | |||
| Ca(HCO3)2(т) | –2344 | H2Se (г) | Fe | ||||
| Ca(OH)2 | –986 | H2Te (г) | H2 | ||||
| CaSO4 | –1424 | KСl (т) | –437 | K | |||
| CaSiO3 | –1584 | KСlO3 (т) | –398 | Mg | |||
| CaO×Al2O3 | –2321 | KСlO4 (т) | –430 | N2 | |||
  | |||||||
| Продолжение табл. 2.1 | |||||||
| Вещество | ∆ Hобр | S 0 | Вещество | ∆ Hобр | S 0 | Вещество | S 0 |
| CaO×SiO2×2H2O | –302 | MgSO4 | –1277 | Na | |||
| CaSO4×0,5H2O | –1575 | Mg(OH)2 (т) | –925 | Ni | |||
| CaSO4×2H2O | –2021 | N2O (г) | O2 | ||||
| 3CaO×Al2O3 | N2O4 (г) | Pb | |||||
| 3CaO×SiO2 | –444 | NaHCO3 (т) | –914 | S | |||
| CH4(г) | –75 | NaOH (т) | –425 | Se | |||
| C2H2(г) | NH3 (г) | –46 | Si | ||||
| C2H5OH(г) | –277 | (NH4)2 Cr2O7 | –1808 | - | Te | ||
| C2H6 | –85 | NO (г) | V | ||||
| C3H8 | –104 | NO2 (г) | Ge | ||||
 C6H5NH2(ж) C6H5NH2(ж) | Ni (OH)2 (т) | –543 | |||||
| C6H6(ж) | NiS | –79 | |||||
| C7H8(г) | NiCl2 | –304 | |||||
| CO(г) | –110 | NiO | –240 | ||||
| CO2(г) | –393 | SiO2 | –859 | ||||
| Cr2O3(т) | –1141 | SO2 (г) | –297 | ||||
| CrO3(т) | –590 | SO3 (г) | –396 | ||||
| Cr(OH)3 | –1014 | SeO2 | –225 | ||||
| Cu2O | –167 | SiH4 | |||||
| CuO | –165 | MgCO3 | –1086 | ||||
| CuS | –49 | PbO (т) | –219 | ||||
| FeCl2 | –342 | PbO2 (т) | –277 | ||||
| Fe3O4 | –1118 | Pb(OH)2 (т) | –513 | ||||
| MgO | –601 | TeO2 | –52 | ||||
| BaCO3 | –1213 | BaSO4 | –1458 |
Таблица 2.2
Стандартные теплоты сгорания
| Вещество | 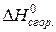 , кДж/моль , кДж/моль |
| CO CH4 C2H2 C2H6 C2H5OH CH3COOH CH3COOC2H5 C2H3COOH H2 NH3 N2O | –283 –890 –1300 –1560 –1367 –874 –2254 –1370 –286 –383 –69 |
Все процессы, протекающие в термодинамических системах, можно разделить на два класса – самопроизвольные и несамопроизвольные. Самопроизвольные процессы всегда протекают необратимо, а несамопроизвольные могут быть обратимыми. Обратимым называют такой процесс, который можно в любой момент заставить протекать в обратном направлении через те же стадии, по которым он протекал в прямом направлении. Обратимые процессы никогда не протекают самопроизвольно, они всегда требуют внешних воздействий (совершения работы и подведения или отведения тепла).
Самопроизвольные процессы в термодинамических системах протекают самопроизвольно, без каких-либо внешних воздействий. Вода сама течёт вниз по склону, теплота переходит от нагретых тел к холодным, а не наоборот. Самопроизвольные процессы, протекая в одном направлении, никогда затем не идут в обратном направлении самопроизвольно, они однонаправленны.
Первое начало термодинамики ничего не говорит о направлении процессов, протекающих в природе и технике. Второе начало (закон) термодинамики даёт такие предсказания. Математическое выражение второго начала термодинамики осуществляется с помощью введения специальной аддитивной функции состояния, которая называется энтропией, малое изменение которой dS по определению равно:
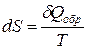 , , | (2.10) |
где Т – абсолютная температура системы. Размерность энтропии – Дж/К. Постулируем основное неравенство, являющееся одним из математических выражений второго начала термодинамики:
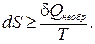 | (2.11) |
Величина 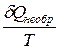 называется приведённой теплотой. Изменение энтропии при протекании процесса в системе больше или равно сумме приведённых теплот в этом процессе.
называется приведённой теплотой. Изменение энтропии при протекании процесса в системе больше или равно сумме приведённых теплот в этом процессе.
Если система замкнута (изолирована), она не обменивается энергией с окружающей средой и неравенство (2.11) приобретает вид (2.12)
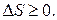 | (2.12) |
Неравенство (2.12) называется законом возрастания энтропии, являющимся наиболее общей формулировкой второго начала термодинамики: энтропия изолированной системы при любых протекающих в ней процессах никогда не убывает, она либо растёт, либо остаётся постоянной. Последнее реализуется, когда система пребывает в состоянии равновесия. При протекании самопроизвольного процесса в замкнутой системе энтропия всегда растёт. Очевидно, энтропия в системе СИ измеряется в Дж/К.
Статистический смысл энтропии. Макроскопическое состояние термодинамической системы задаётся её макроскопическими переменными, такими как объём, давление, температура, концентрации и другие. Если для двух состояний системы эти переменные одинаковы, то и состояния одинаковы. Однако при микроскопическом рассмотрении системы легко обнаруживается, что данному макроскопическому состоянию системы соответствует весьма большое число неотличимых с макроскопической точки зрения микроскопических состояний. Так, например, данной величине давления газа на стенку сосуда соответствуют в разные моменты времени разные положения различных молекул по отношению друг к другу и удары молекул о стенку сосуда. Можно говорить о неком, при этом весьма большом, среднем числе W микросостояний системы, соответствующих данному макроскопическому состоянию. Больцман показал, что абсолютная энтропия системы связана с логарифмом среднего числа микросостояний системы W:
| S = k lnW, | (2.13) |
где k – константа Больцмана, k = 1,381·10-23 Дж/К. Из формулы (2.13) видно, что энтропия – мера статистического беспорядка в системе. При переходе вещества от кристаллического состояния к жидкому, а затем от жидкости к газу, его энтропия растёт, значит, растёт и число микросостояний, последовательно отвечающих каждому из этих макроскопических состояний. Учитывая статистический смысл энтропии, второе начало термодинамики можно сформулировать следующим образом: изолированная система, находящаяся в неравновесном состоянии, стремится к состоянию равновесия, которому отвечает наибольшее среднее число микросостояний системы.
Для того чтобы выбрать начало отсчета абсолютной энтропии термодинамической системы, используют третье начало термодинамики, которое формулируется согласно Планку следующим образом: энтропия всех индивидуальных веществ, существующих в виде идеальных кристаллов при температуре абсолютного нуля, равна нулю.
Предположим, что температура одного моля индивидуального вещества при постоянном давлении, например стандартном, увеличивается от абсолютного нуля (где вещество находится в состоянии идеального кристалла) до температуры Т, где вещество находится в газообразном состоянии. Тогда осуществляются следующие процессы нагревания вещества и его переходов из одних агрегатных состояний в другие.
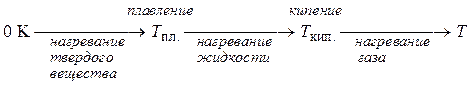
Эти процессы могут протекать обратимо.
Энтропия в конечном состоянии с температурой Т равна сумме изменений энтропии, произошедших на всех предыдущих этапах. Определенная таким образом величина S(T) есть абсолютная энтропия одного моля индивидуального вещества при температуре T и давлении P, выражается в Дж/(К·моль). В термохимических таблицах обычно приводятся стандартные мольные абсолютные энтропии индивидуальных веществ S 0, определяемые для стандартных условий (Т = 298,15 К, Р = 101325 Па) (см. табл. 2.1).
Поскольку энтропия – аддитивная функция состояния, расчет изменения энтропии химической реакции 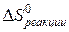 , протекающей до конца, проводится аналогично расчету энтальпии химической реакции
, протекающей до конца, проводится аналогично расчету энтальпии химической реакции 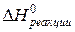 :
:
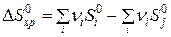 , , | (2.14) |
где 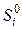 ,
, 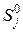 – абсолютные мольные энтропии продуктов реакции и исходных веществ соответственно в Дж/(К·моль).
– абсолютные мольные энтропии продуктов реакции и исходных веществ соответственно в Дж/(К·моль).
В уравнении (2.14), в отличие от подсчета величины 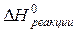 по (2.8), следует учитывать стандартные мольные энтропии как сложных, так и простых веществ. Простым веществам невозможно приписать нулевые значения стандартных энтропий.
по (2.8), следует учитывать стандартные мольные энтропии как сложных, так и простых веществ. Простым веществам невозможно приписать нулевые значения стандартных энтропий.
В случае закрытой системы постоянного состава, которая может совершать только работу расширения, можно объединить I и II начала термодинамики для обратимого процесса. Для этого в уравнение (2.2) подставим величину 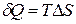 из уравнения (2.9) и получим (2.15):
из уравнения (2.9) и получим (2.15):
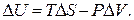 | (2.15) |
Это уравнение часто называют фундаментальным уравнением для закрытой системы постоянного состава.
Приведем некоторые важные положения термодинамики без доказательства. Вводится функция состояния G термодинамической системы, определяемая равенством 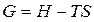 . Функция G называется изобарно-изотермическим потенциалом, или просто изобарным потенциалом, или потенциалом Гиббса, измеряется в джоулях. В изобарно-изотермических условиях для закрытой термодинамической системы имеется важный критерий самопроизвольности протекания процессов:
. Функция G называется изобарно-изотермическим потенциалом, или просто изобарным потенциалом, или потенциалом Гиббса, измеряется в джоулях. В изобарно-изотермических условиях для закрытой термодинамической системы имеется важный критерий самопроизвольности протекания процессов:
| ∆G = ∆H – T∆S, | (2.16) |
где ∆G – изменение изобарно-изотермического потенциала или потенциала Гиббса при протекании некоторого процесса в термодинамической системе. Критерий протекания самопроизвольных процессов: процесс в термодинамической системе протекает самопроизвольно при постоянных давлении и температуре, если изменение изобарного потенциала в системе меньше нуля (∆G < 0).
Если ∆G = 0, то система находится в состоянии равновесия;
если ∆G > 0, то протекает несамопроизвольный процесс. Часто вместо величины ∆G процесса, связанного с протеканием химической реакции, вычисляют величину ∆G 0, значение которой определяется равенством (2.17):
| ∆G 0 = ∆H 0 – T∆S 0. | (2.17) |
Величина ∆G 0 вычисляется так, как если бы химическая реакция протекала до конца. Величины ∆Н 0 и ∆S 0 вычисляются по формулам (2.8) и (2.14). Важность величины ∆G 0 заключается в том, что она связана с величиной константы химического равновесия K выражением (2.18):
| ∆G 0 = –RT ln K. | (2.18) |
Отметим, что в выражении (2.18) константа равновесия K безразмерна. Эта величина выражается или через парциальные давления реагентов, деленные на стандартное давление, или через концентрации реагентов, деленные на стандартные концентрации (например, 1 моль/л). Выражение (2.18), если вычислена величина ∆G 0для конкретной реакции, позволяет судить о величине константы равновесия и о том, в сторону какой реакции сдвинуто равновесие (K < 1 – в сторону исходных реагентов, K > 1 – в сторону продуктов реакции, K = 1 – равновесие примерно посередине).
Пример 3. Рассмотрите протекание реакции разложения перхлората калия в закрытой системе:
4KClO4(тв) = 2KClO3(тв) + 2KCl(г) + 5O2(г); ∆ H0хр = 50 кДж.

