 2015-03-22
2015-03-22 15871
15871Итак, как нами было выяснено выше, получаемые в ходе решения вариационной задачи Релея – Ритца для молекулярных систем, получают решения, отвечающие молекулярным орбиталям трёх типов – связывающие, разрыхляющие и несвязывающие. При этом в зависимости от типа комбинируемых атомных орбиталей, различают молекулярные орбитали 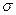 - и
- и 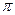 - типа. Орбитали обоих типов могут быть как связывающими, так и разрыхляющими. Поглощение света сопровождается переходом электронов со связывающей - или - орбитали на разрыхляющую
- типа. Орбитали обоих типов могут быть как связывающими, так и разрыхляющими. Поглощение света сопровождается переходом электронов со связывающей - или - орбитали на разрыхляющую 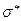 или
или 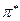 - орбитали. В связи с этим различают
- орбитали. В связи с этим различают 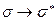 и
и 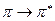 переходы. Не участвующие в образовании химических связей электроны неподелённых электронных пар образуют несвязывающие
переходы. Не участвующие в образовании химических связей электроны неподелённых электронных пар образуют несвязывающие 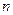 - орбитали. Энергия неподелённой пары электронов в молекуле равна её энергии в изолированном атоме. Несвязываюшие - орбитали локализованы на отдельных атомах, тогда, как - и - орбитали обычно распространяются на два или более атома в молекуле. Как видно из приведенной выше схемы, уровень несвязывающей молекулярной орбитали расположен выше уровней связывающих - и - орбиталей. В основном состоянии -, - и - орбитали обычно заняты электронами, а - и - орбитали свободны. Поглощение света происходит в результате переходов с занятых орбиталей на свободные.
- орбитали. Энергия неподелённой пары электронов в молекуле равна её энергии в изолированном атоме. Несвязываюшие - орбитали локализованы на отдельных атомах, тогда, как - и - орбитали обычно распространяются на два или более атома в молекуле. Как видно из приведенной выше схемы, уровень несвязывающей молекулярной орбитали расположен выше уровней связывающих - и - орбиталей. В основном состоянии -, - и - орбитали обычно заняты электронами, а - и - орбитали свободны. Поглощение света происходит в результате переходов с занятых орбиталей на свободные.

Рис. 30. Типы электронных переходов.
Наибольшей энергии, очевидно, будет требовать переход, характерный для насыщенных молекул и соответствующих поглощению в вакуумном УФ (l<200 нм). Переходы типа происходят в молекулах с сопряжёнными связями и в ароматических молекулах. Они связаны с поглощением квантов энергии соответственно в видимой или ближней УФ – областях спектра. Переходы 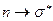 и
и 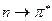 обычно происходят в молекулах, содержащих гетероатомы (например, атомы N, O, S и т.д.).
обычно происходят в молекулах, содержащих гетероатомы (например, атомы N, O, S и т.д.).
Хромофоры – это функциональные группы, которые поглощают электромагнитное излучение независимо от того, возникает при этом окраска или нет. Они содержат группировки атомов, содержащие - электроны или свободные электронные пары гетероатомов, которые дают свои, характеристические линии поглощения в УФ – области спектра.
Ауксохромы – это функциональные группы, например, 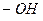 ,
, 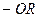 ,
, 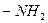 и другие, которые, вступая в сопряжение с хромофором за счёт своих неподелённых электронов, становятся частью нового, более протяжённого хромофора.
и другие, которые, вступая в сопряжение с хромофором за счёт своих неподелённых электронов, становятся частью нового, более протяжённого хромофора.
В некоторых случаях на интенсивность и положение полосы поглощения влияют природа растворителя или окружение хромофора, причём в результате полоса может сдвигаться как в длинноволновую, так и в коротковолновую область.
Таблица 28. Типичные хромофоры и их характеристики.
| Хромофор | Длина волны, нм | Коэффициент молярной экстинкции (eмах) и интенсивность полосы поглощения | Возбуждение |
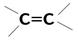 | 14 000 сильная 8 000 сильная | p-электронов | |
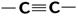 | 10 000 сильная 2 000 сильная 150 слабая | p-электронов | |
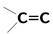 | 18 000 сильная 5 000 сильная 15 слабая | p-электронов Свободной электронной пары кислорода | |
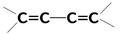 | 20 000 сильная | p-p* | |
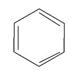 | 60 000 сильная 4 400 средняя 204 слабая | p-p* n-p* | |
 | Полоса средней силы | Свободной электронной пары кислорода | |
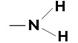 | Полоса средней силы | Свободной электронной пары азота | |
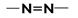 | 340 – 370 | Слабая полоса | Свободных электронных пар азота |
Согласно старой номенклатуре, могут иметь место следующие эффекты:
1. Батохромный сдвиг (или красный сдвиг) – в сторону длинных волн. Такой сдвиг могут вызывать, например, алкильные группы, расположенные по соседству с хромофором;
2. Гипсохромный сдвиг (или синий сдвиг) – в сторону коротких волн;
3. Гиперхромный эффект – повышение интенсивности поглощения;
4. Гипохромный эффект – понижение интенсивности поглощения.
Применение УФ – спектроскопии для идентификации и анализа полимеров ограничено соединениями, содержащими хромофорные группы. Это в частности полиены, ароматические полимеры, содержащие - связи и особенно сопряжённые системы, а также полимеры с карбонильными группами. Характеристические линии поглощения многих ароматических (бензол, нафталин) и гетероароматических соединений (пиридин, хинолин) зависят от протяжённости и расположения -электронной системы. Это позволяет распознавать различные соединения одного и того же ряда. Метод УФ – спектроскопии используют также для определения содержания в полимерах различных примесей следов веществ, дающих характеристические линии поглощения в исследуемой области УФ – спектра. Остановимся теперь на проблеме возможности применения неэмпирических и полуэмпирических расчётных схем частот и вероятностей электронных переходов, которые имеют место в молекулах сопряжённых системах при поглощении последними квантов электромагнитной энергии. Квантовохимические расчёты геометрии и распределения электронной плотности для возбуждённых состояний, выполненные любым методом, представляют интерес, так как здесь даже полуколичественные результаты оказываются весьма полезными. Однако при вычислении параметров электронных спектров поглощения требования к точности расчёта выше, поэтому для этой цели в настоящее время используются, несмотря на трудоёмкость, практически только различные варианты метода конфигурационного взаимодействия (КВ). Для изучения электронных спектров поглощения молекул, содержащих системы сопряженных - связей, наиболее широко используются расчёты полуэмпирическим методом ППДП/С с учётом KB. Предложено много модификаций этого метода, которые немного отличаются друг от друга за счёт выбора эмпирических параметров, но приводят к довольно близким результатам. Ошибки при практических расчётах энергии электронных переходов - типа даже для довольно сложных молекул не превышают нескольких десятых электрон-вольт. Для электронных переходов - типа, расхождение данных расчёта и эксперимента больше, причём вычисленные значения в большинстве случаев получаются заниженными. Последнее, по-видимому, связано с использованием приближения ППДП, в котором не учитывается ряд интегралов взаимодействия между электронами, вносящих существенный вклад в энергии - переходов. При этом синглет – триплетное расщепление для - возбуждённых состояний получается равным нулю, хотя оно в действительности для некоторых соединений достигает 1 эВ. Этот недостаток легко устранить, перейдя к расчётам в приближениях ЧПДП и ПДДП. Однако расчёты электронных спектров поглощения в этих приближениях широкого распространения не получили. Для расчёта энергий переходов - типа полуэмпирические методы применялись мало, точность при использовании существующих параметризаций должна быть невысокой, и можно ожидать лишь весьма грубых качественных результатов. Следует также напомнить, что в полуэмпирических методах обычно используется минимальный слэйтеровский базис и это не позволяет рассчитать параметры переходов Ридберга, которые обычно лежат в области электронных переходов - типа и могут смешиваться с ними. Для того чтобы иметь возможность рассчитать параметры ридберговских переходов полуэмпирическими методами, необходимо расширить базис, включив в него атомные орбитали с большими квантовыми числами. Работы в этой области проводились рядом авторов, но к успеху не привели. Оказалось, что с помощью полуэмпирических методов удаётся получить удовлетворительное согласие с экспериментом для энергий ридберговских переходов, но их отнесение часто не совпадает с данными более надёжных неэмпирических расчётов. Опубликовано много работ, в которых параметры электронно-возбужденных состояний и электронных спектров поглощения вычислялись неэмпирическими методами. В них было показано, что хорошее согласие с экспериментом удаётся получить лишь при использовании базисов очень большого размера. Попытки подобрать небольшие наборы базисных орбиталей для расчёта электронных спектров поглощения кончились безрезультатно. В небольшом базисе удавалось получить согласие с экспериментом только для отдельных электронных переходов для ограниченного круга соединений. Это связано с тем, что изменение распределения электронной плотности при возбуждении нельзя передать с необходимой степенью точности простым варьированием заселённостей молекулярных орбиталей и даже переходом к их линейным комбинациям. Иначе говоря, набор базисных орбиталей подобранный для основного состояния, оказывается неудовлетворительным для возбужденного. При возбуждении электронное облако обычно расширяется, и это сильно сказывается на величине матричных элементов взаимодействия электронов между собой и с атомными ядрами. Для того чтобы волновая функция, представленная в виде линейной комбинации базисных орбиталей могла передать это расширение, базис, подобранный для расчёта параметров основного состояния, необходимо модифицировать. Это можно сделать, изменив показатели орбитальных экспонент или включив дополнительно диффузные орбитали. В первом случае размер базиса не увеличивается, но расчёт не универсален, так как характер расширения электронного облака специфичен для каждого электронного перехода. Второй способ (расширение базиса) позволяет передать изменение волновой функции при любом электронном переходе, поэтому он является единственным рациональным. Однако для получения хорошего согласия с экспериментом одного очень большого базиса мало при неэмпирическом расчёте электронных спектров поглощения. Необходимо еще учесть взаимодействие с очень большим числом электронно-возбужденных конфигураций. Обычно эти расчёты проводят по следующей методике: делают предварительную оценку по теории возмущений и все однократно и дважды возбужденные конфигурации разделяют на три группы (вводят два пороговых параметра). К первой группе относят конфигурации, которые вносят заметный вклад в электронную волновую функцию возбужденного состояния, взаимодействие с ними учитывают методом конфигурационного взаимодействия; ко второй группе относят конфигурации, которые вносят средний вклад в электронную волновую функцию возбужденного состояния, взаимодействие с ними учитывают по теории возмущений; к третьей группе относят конфигурации, которые вносят малый вклад в электронные волновые функции возбужденных состояний, взаимодействием с ними пренебрегают. Однако расчёты по такой методике в настоящее время можно проделать лишь для небольших молекул, так как с увеличением размера соединения трудности вычислений очень быстро возрастают.
Неэмпирические методы наиболее широко применяются для расчёта энергии ридберговских переходов. Для несложных молекул удаётся вычислить положение полос поглощения, однако выводы об их отнесении не всегда достоверны, так как полосы Ридберга лежат обычно очень близко друг от друга. Даже у трёх - пяти атомных молекул в области 7 - 8 эВ может быть расположено до десяти ридберговских переходов. Для соединений с атомами тяжёлых элементов отнесение переходов к ридберговским на основе данных аналогичных расчётов является сомнительным. Это связано с уменьшением различий между валентными орбиталями, а также орбиталями, с большими значениями главного квантового числа. В этом случае из анализа распределения электронной плотности в основном и электронно-возбужденных состояниях очень трудно, а для многих соединений практически невозможно дать достоверный ответ на вопрос, за счёт какого фактора произошло расширение электронного облака – за счёт изменения валентных орбиталей или за счёт заселения орбиталей с большими квантовыми числами. Для энергий нижних - электронных переходов неэмпирические расчеты приводят к завышенным величинам, ошибки обычно превышают 1 эВ. В работе на примере молекулы этилена было показано, что для получения хорошего согласия с экспериментом для энергии - электронного перехода необходимо очень сильно расширять базис. По-видимому, это связано с тем, что мы еще плохо представляем себе, как изменяются волновые функции при - электронных переходах, и не знаем, как можно передать это изменение с помощью относительно небольшого базиса. Подобные трудности ранее возникали при проведении неэмпирических расчётов термохимических параметров анионов, но в настоящее время они преодолены за счёт подбора специальных наборов базисных орбиталей. Практика расчётов электронных спектров поглощения органических соединений распространяется на область не далее 7 - 10 эВ. Для более далёкой области вычисления не производят из-за значительного возрастания трудностей, которые связаны в первую очередь с необходимостью учёта взаимодействия между очень большим числом высоковозбужденных конфигураций. Силы осцилляторов электронных переходов во всех методах рассчитываются менее точно, чем их энергии, и результаты могут отличаться от данных эксперимента в несколько раз. Требования к точности волновых функций при этом особенно велики, и небольшие погрешности, связанные с выбором базиса и отбором конфигураций, могут сильно отразиться на результатах. Однако силы осциллятора электронных переходов, наблюдаемых экспериментально, различаются на несколько порядков. В этих условиях даже полуколичественные оценки достаточны для предсказания общего вида спектра поглощения и для его интерпретации (отнесения отдельных полос к электронным переходам определенного типа). Таким образом, видно, что результаты расчёта электронных спектров поглощения можно использовать для интерпретации экспериментальных данных и извлечения из них интересной информации о строении сложных молекул. Однако делать это следует очень осторожно, всегда помня о приближенном характере вычислений. Особую осторожность необходимо проявлять при использовании вычисленных сил осцилляторов. Рассматривая - электронные переходы (видимая и ближняя ультрафиолетовые области спектра), следует пользоваться полуэмпирическими методами, так как они дают более точные результаты. Для интерпретации экспериментальных данных о ридберговских переходах надо пользоваться неэмпирическими расчетами. В настоящее время экспериментальная техника позволяет регистрировать электронные спектры поглощения в очень далекой УФ – области. Практики квантово – механических расчётов для переходов с такой большой энергией нет. Однако, судя по косвенным данным, удовлетворительные полуколичественные результаты можно получить из результатов квантово – механических расчётов и для этой области. Мы пришли к такому заключению на основе анализа результатов квантово – механических вычислений электронных поляризуемостей и гиперполяризуемостей органических молекул. Величина этих параметров определяется значениями энергий и сил осцилляторов всей совокупности электронных переходов. Хорошее согласие с экспериментом, полученное для них многими авторами, косвенно свидетельствует, что квантово – механических расчёт приблизительно правильно передаёт электронный спектр поглощения и в дальней ультрафиолетовой области спектра.

