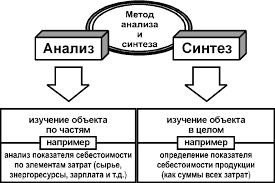
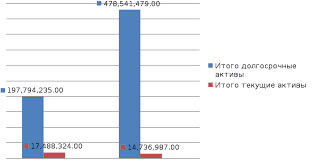
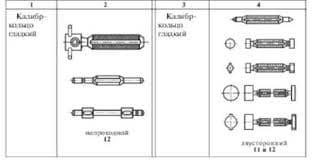
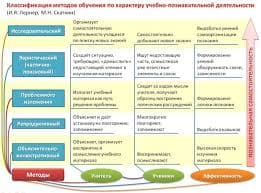
2015-03-22
2015-03-22 836
836палительной природы (геморрагические лихорадки, иммунные васкулиты), а также при ги-повитаминозах (цинга и др.). В особую подгруппу выделяют вазопатии, связанные с генетически обусловленным истончением субэндотелия и недоразвитием его коллагенового каркаса, в результате чего возникают легко кровоточащие аневризмы мелких сосудов (телеангиэктазы) и сосудистые шунты.
2) Геморрагические диатезы и синдромы,
обусловленные нарушениями тромбоцитарно-
го гемостаза. К ним относятся формы с низким
содержанием в крови тромбоцитов (тромбоцито-
пении) и с нарушениями структуры и функции
тромбоцитов (тромбоцитопатии). Как уже ука
зывалось выше, последние могут быть связаны
с отсутствием или неправильной функцией ре
цепторов мембран тромбоцитов, с малым содер
жанием в этих клетках плотных и альфа-гра
нул, а также с нарушением их высвобождения,
с блокадой образования тромбоксана А2, с нару
шениями транспорта в тромбоциты кальция и с
другими причинами. В особую группу выделя
ют ятрогенные (лекарственные) тромбоцитопа
тии, вызываемые аспирином и другими антиаг-
регантами, антагонистами транспорта ионов
кальция, блокаторами рецепторов и др.
Симптоматические (вторичные) тромбоцитопатии и тромбоцитопении наблюдаются при ги-попластической и В12-дефицитной анемиях, при острых лейкозах, уремии, лучевой болезни, при некоторых эндокринных заболеваниях, отравлениях и гепатолиенальном синдроме, особенно протекающем с портальной гипертензией.
3) Нарушения свертываемости крови (коа-
гулопатии). В эту группу включаются наслед
ственные коагулопатии, среди которых домини
руют (около 97%) гемофилии А и В, а также
болезнь Виллебранда, при которой нарушение
адгезивности и ристомицин-агрегации тромбоци
тов часто сочетается со снижением активности
фактора VIII.
В основе обоих видов гемофилии лежит мутация локусов синтеза фактора VIII (гемофилия А) или фактора IX (гемофилия В) в Х-хромосо-ме. Болезнь при обеих формах наследуется по рецессивному, сцепленному с полом типу; носителями болезни являются женщины, а больными - только лица мужского пола (исключения из этого правила крайне редки). Все дочери больного гемофилией, получившие патологическую
Часть III. ПАТОФИЗИОЛОГИЯ ОРГАНОВ И СИСТЕМ
Х-хромосому от отца, являются кондукторами болезни, а сыновья женщин-носительниц гемофилии в 50% случаев имеют шанс стать больными, дочери - носителями патологического гена.
Для гемофилии характерна кровоточивость гематогенного типа - очень болезненные кровоизлияния в крупные суставы, мышцы, забрю-шинную клетчатку, в область черепа, гематурия, тяжелые посттравматические и послеоперационные кровотечения, в том числе при малых травмах и вмешательствах (порезы, удаления зубов и т. п.).
Поскольку факторы VIII и IX участвуют только во внутреннем механизме свертывания крови, при гемофилии удлинены общее время свертывания цельной крови, время свертывания ре-кальцифицированной цитратной плазмы и АПТВ, тогда как протромбиновыи показатель и тромбиновое время свертывания остаются нормальными.
В отличие от наследственных коагулопатий большинство приобретенных форм этой патологии связано с нарушениями синтеза факторов протромбинового комплекса - факторов II, V, VII и X. Поскольку все эти факторы синтезируются в печени, их дефицит наблюдается при тяжелых заболеваниях этого органа, а также при нарушениях всасывания в кишечнике витамина К (обтурационная желтуха, кишечный дис-бактериоз), при приеме антагонистов витамина К. Аналогичный вид кровоточивости наблюдается при геморрагической болезни новорожденных. Для всех этих форм характерно преимущественное замедление свертывания в протром-биновом тесте при одновременном снижении показателей АПТВ (из-за депрессии факторов II, IX и X) при нормальном тромбиновом времени свертывания.