2015-05-30
2015-05-30 17938
17938Фармакокинетика — раздел клинической фармакологии, предметом которого является изучение процессов всасывания, распределения, связывания с белками, биотрансформации и выведения лекарственных веществ. Ее развитие стало возможным благодаря разработке и внедрению в практику высокочувствительных методов определения содержания лекарственных веществ в биологических средах — газожидкостной хроматографии, радиоиммунных, ферментно-химических и других методов, а также благодаря разработке методов математического моделирования фармакокинетических процессов. На основании данных о фармакокинетике того или иного препарата определяют дозы, оптимальный путь введения, режим применения препарата и продолжительность лечения. Регулярный контроль содержания лекарственных средств в биологических жидкостях позволяет своевременно корригировать лечение.
Фармакокинетические исследования необходимы при разработке новых препаратов, их лекарственных форм, а также при экспериментальных и клинических испытаниях ЛС.
Процессы, происходящие с лекарственными препаратами в организме, могут быть описаны с помощью ряда параметров.
Одним из основных показателей, определяющих фармакологический эффект, считают концентрацию ЛС на уровне рецептора, однако в условиях целостного организма установить её невозможно. В эксперименте было доказано, что в большинстве случаев между концентрацией препарата в крови и его содержанием в области рецептора существует корреляция.
В связи с этим для определения фармакокинетических параметров изучают содержание ЛС в крови. Для того чтобы получить соответствующее представление о поступлении препарата в кровь и выведении его из организма, отслеживают изменения концентрации ЛС в плазме крови на протяжении длительного времени. Содержание препаратов в плазме крови определяют методами жидкостной или газожидкостной хроматографии, с помощью радиоиммунного или иммуноферментного анализа и другими способами.
На основании полученных данных строят график. На оси абсцисс отмечают время от начала исследования, а на оси ординат - концентрацию ЛС в плазме крови (в соответствующих единицах).
Такой график носит название фармакокинетической кривой (рис. 1).
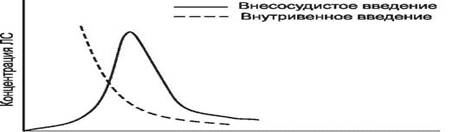 Время после введения
Время после введения
Концентрация лекарственного вещества (С) -это ее количество в определенном объеме крови в конкретный момент времени после введения в организм.
Константы скорости элиминации (Кel), абсорбции (Ка) и экскреции (Кex) – характеризуют соответственно скорость исчезновения препарата из организма путем биотрансформации и выведения, скорость поступления его из места введения в кровь и скорость выведения с мочой, калом, слюной и др.
Период полувыведения (Т1/2) — время, необходимое для уменьшения вдвое концентрации препарата в крови, зависит от константы скорости элиминации (Т1/2= 0,693/Кel).
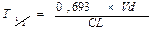
где Т1/2 – период полувыведения; 0,693 – коэффициент, который является логарифмом от 2; Vd - объем распределения; Сl - общий клиренс.
Константа элиминации (Кel) - процент уменьшения концентрации ЛВ в крови за единицу времени. Чем больше Кel, тем быстрее ЛВ выводится из крови. Константа элиминации зависит от периода полувыведения:
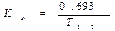
Период полуабсорбции (Т1/2а) - время, необходимое для всасывания половины дозы препарата из места введения в системный кровоток; пропорционален скорости абсорбции (Т1/2а = 0,693/Ка).
Константа абсорбции (Ка) - характеризует скорость всасывания ЛВ в организме человека или животного. Константа абсорбции зависит от периода полувыведения:
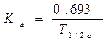
Распределение препарата в организме характеризуют период полураспределения, кажущаяся начальная и стационарная (равновесная) концентрации, объем распределения.
Период полураспределения (Т1/2,a) — время, необходимое для достижения концентрации препарата в крови, равной 50% от равновесной, т.е. при наличии равновесия между кровью и тканями.
Кажущаяся начальная концентрация (С0) — концентрация препарата, которая была бы достигнута в плазме крови при внутривенном его введении и мгновенном распределении по органам и тканям.
Равновесная концентрация (Сss) — концентрация препарата, которая установится в плазме (сыворотке) крови при поступлении препарата в организм с постоянной скоростью. При прерывистом введении (приеме) препарата через одинаковые промежутки времени в одинаковых дозах выделяют максимальную (Сssmax) и минимальную (Сssmin) равновесные концентрации.
Объем распределения препарата (Vd - volume of distribution) характеризует степень его захвата тканями из плазмы (сыворотки) крови. Vd (Vd= D/C0) — условный объем жидкости, в котором нужно растворить всю попавшую в организм дозу препарата (D), чтобы получилась концентрация, равная кажущейся начальной концентрации в сыворотке крови (С0).
Общий клиренс препарата (Clt) характеризует скорость “очищения” организма от лекарственного препарата.
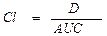 где Сl – общий клиренс; D – доза введенного препарата; AUC – площадь под фармакокинетической кривой.Выделяют почечный (Clr) и внепочечный (Cler) клиренсы, которые отражают выведение лекарственного вещества соответственно с мочой и другими путями (прежде всего с желчью). Общий клиренс является суммой почечного и внепочечного клиренса.
где Сl – общий клиренс; D – доза введенного препарата; AUC – площадь под фармакокинетической кривой.Выделяют почечный (Clr) и внепочечный (Cler) клиренсы, которые отражают выведение лекарственного вещества соответственно с мочой и другими путями (прежде всего с желчью). Общий клиренс является суммой почечного и внепочечного клиренса. 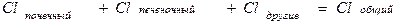
Площадь под кривой “концентрация — время” (AUC - area under the curve)— площадь фигуры, ограниченной фармакокинетической кривой и осями координат (AUC = C0/Kel). Величина (AUC) связана с другими фармакокинетическими параметрами — объемом распределения, общим клиренсом. При линейности кинетики препарата в организме величина AUC пропорциональна общему количеству (дозе) препарата, попавшего в системный кровоток. Часто определяют площадь под частью кривой (от нуля до некоторого времени t); этот параметр обозначают AUCt, например, площадь под кривой от 0 до 8 ч — AUC8.
Абсолютная биодоступность (f) — часть дозы препарата (в %), которая достигла системного кровотока после внесосудистого введения, равна отношению AUC после введения исследуемым методом (внутрь, в мышцу и др.) к AUC после внутривенного введения. Относительную биодоступность определяют для сравнения биодоступности двух лекарственных форм для внесосудистого введения. Она равна отношению (AUC’/AUC)(D/D’) после введения двух сравниваемых форм. Общая биодоступность — часть принятой внутрь дозы препарата, которая достигла системного кровотока в неизмененном виде и в виде метаболитов, образовавшихся в процессе всасывания в результате так называемого пресистемного метаболизма, или “эффекта первичного прохождения”.
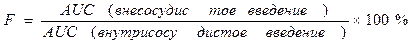
Биоэквивалентность (сравнительная биодоступность) - это соотношение количества ЛВ, поступающего в кровь при введении его в различных лекарственных формах (или ЛС разных фирм). Если лекарственные препараты демонстрируют схожую биодоступность, они расцениваются как биоэквивалентны.
ВСАСЫВАНИЕ — процесс поступления лекарственного вещества из места введения в кровь. Существуют четыре механизма всасывания ЛС при энтеральном введении (рис. 2):
Ø пассивная диффузия;
Ø активный транспорт;
Ø фильтрация через поры;
Ø пиноцитоз
Прохождение большинства лекарственных препаратов через слизистую оболочку пищеварительного тракта определяется их растворимостью в липидах и ионизацией. При приеме лекарственных веществ внутрь скорость их абсорбции отличается в различных отделах ЖКТ.
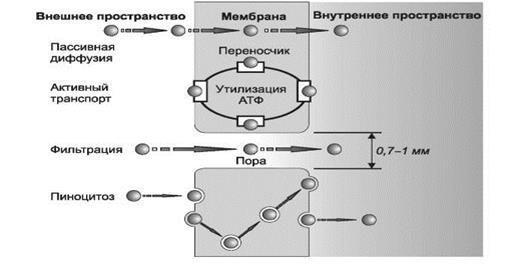
Q - молекула лекарственного вещества
После прохождения через стенку желудка и/или кишечника лекарственный препарат поступает в печень. Некоторые лекарственные вещества под влиянием ферментов печени подвергаются значительным изменениям (“эффект первичного прохождения”). Именно поэтому, а не вследствие плохой абсорбции, для достижения достаточного эффекта дозы некоторых препаратов (пропранолола, аминазина, опиатов) при приеме их внутрь должны быть значительно больше, чем при внутривенном введении. Биотрансформацию вещества при первичном прохождении через печень в процессе всасывания называют пресистемным метаболизмом. Интенсивность пресистемного метаболизма зависит от скорости тока крови в печени.
На процесс всасывания лекарств в желудке и кишечнике оказывает влияние рН, который в желудке равен 1-3, в двенадцатиперстной кишке — 5-6, а в тонкой и толстой кишках — около 8. Кислоты легче всасываются в желудке, а основания — в тонкой или толстой кишке.
Под действием кислой среды желудка некоторые лекарственные средства, в частности бензилпенициллин, могут разрушаться.
На лекарственные препараты оказывают также действие ферменты желудочно-кишечного тракта, которые способны инактивировать белки и полипептиды (АКТГ, вазопрессин, инсулин и т.д.), а также некоторые другие вещества (прогестерон, тестостерон, альдостерон). Соли желчных кислот в свою очередь могут ускорить всасывание лекарственных средств или замедлить его при образовании нерастворимых соединений.
На всасывание лекарственных веществ влияют также моторика желудочно-кишечного тракта, объем и состав пищи, количество принимаемой жидкости, интервал времени между едой и приемом препаратов. Так, молоко нарушает всасывание тетрациклинов, ампициллина и амоксициллина. Следует учитывать и стимулирующее действие пищи на секрецию желудочного сока и соляной кислоты.
Для переноса веществ в ЖКТ особое значение имеют большая площадь поверхности кишечника и влияние постоянного кровотока в слизистой оболочке на градиенты концентрации между просветом кишечника и кровью. Путем диффузии и осмоса через слизистую оболочку кишечника переносятся, в частности, вода, С1 ¯, а также такие вещества, как аскорбиновая кислота, пиридоксин и рибофлавин. Поскольку клеточные мембраны содержат большое количество липидов, для диффузии через мембрану вещества должны быть в некоторой степени жирорастворимыми. Согласно теории неионной диффузии, указанным путем переносятся главным образом недиссоциированные соли слабых кислот или слабых оснований. Это необходимо учитывать при назначении лекарств, большая часть которых всасывается путем диффузии. Для переноса какого-либо вещества в соответствии с уравнением Гендерсона-Гассельбаха особое значение имеет рКа этого вещества и рН в просвете кишечника:
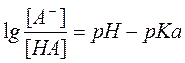 ,
, 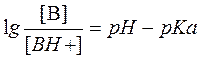 , где
, где
[А¯], [ВН+] – молярные концентрации ионизированных,
[НА], [В] – неионизированных форм кислоты НА и основы В;
рН – кислотно-основной показатель среды;
рКа – логарифм константы диссоциации соединения, количественно равный значению рН, при котором анализируемое соединение диссоциирует наполовину.
Из уравнения видно, что с увеличением значения рН среды диссоциация кислот увеличивается, а оснований - уменьшается.
Таким образом, факторы, влияющие на процессы всасывания ЛВ, разнообразны: растворимость вещества в липидах, степень ионизации молекулы (чем меньше ионизированная молекула, тем лучше она всасывается), перистальтика кишечника, характер и количество пищевой массы, особенности регионарного кровотока, состояние соединительной ткани, агрегантное состояние веществ, сочетание лекарственных средств.