2015-06-05
2015-06-05 3571
3571Среди литейных процессов центральное место занимает переход сплава из жидкого в твердое кристаллическое состояние. Этот процесс является фазовым переходом первого рода, связанным с изменением объема, теплосодержания и энтропии. При рассмотрении данного процесса можно выделить два аспекта: количественный и качественный. Количественный аспект составляет содержание теории затвердевания, главной задачей которой является разработка методов расчета кинетики выделения твердой фазы в объеме расплава. Качественный аспект направлен на рассмотрение вопросов формирования кристаллической структуры отливок и методов управления ею.
ГЛАВА 2.1. ОСНОВНЫЕ ЗАКОНОМЕРНОСТИ САМОПРОИЗВОЛЬНОЙ КРИСТАЛЛИЗАЦИИ МЕТАЛЛОВ И СПЛАВОВ
Современная теория формирования кристаллической структуры отливок.создана за последние 20 лет на основе творческого распространения элементарной теории кристаллизации металлов и сплавов на большие объемы неизотермически затвердевающих расплавов.
Жидкий металл переходит в твердое состояние в соответствии с общими законами термодинамики. Как известно, самопроизвольно протекают те процессы, в которых уменьшается свободная энергия и энергия Гиббса G. На рис. 2.1 приведены кривые изменения энергии Гиббса твердого и жидкого металла в зависимости от изменения температуры. Видно, что при температуре, большей Тпл, Gж < Gтв и устойчивым является жидкое состояние металла. При T < Tпл Gтв < Gж, поэтому устойчивым является твердое состояние. Температура Т = Tпл, при которой Gтв = Gж, называется температурой плавления. При равновесном процессе перехода из жидкого состояния в твердое эта температура соответствует равновесной температуре кристаллизации сплава Tкр. "При снижении температуры ниже Ткр возникает переохлаждение Δ T = Tкр — Т. Так как при этом Gж > Gтв, то создаются термодинамические предпосылки для перехода из жидкого состояния в твердое, т. е. для кристаллизации. Движущей силой этого процесса будет разность энергий Гиббса Δ G = Gж — Gтв. Преобразуем это выражение, прибавив и отняв энергии Гиббса жидкой и твердой фаз при T = Ткр.
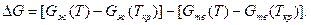
Так как Gж(Ткp) = Gтв(Ткр), то равенство при этой операции не нарушится. Поскольку реальные переохлаждения металлов не велики, то для стоящих в квадратных скобках выра-
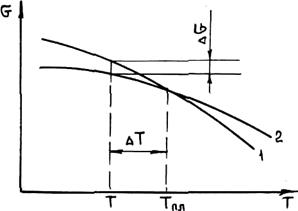
Рис. 2.1. Зависимость энергии Гиббса жидкого (/) и твердого (2) металла от температуры
жений можно написать следующие приближенные равенства:
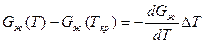 ,
,
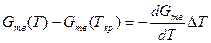 .
.
С учетом этого Δ G = — Δ Т(dGж/dТ — dGтв/dТ). Так как dG/dТ = —S, то Δ G = + Δ T×ΔS, где Δ S — изменение энтропии при плавлении металла. Как известно, Δ S = L/Tпл, где L— молярная теплота плавления, следовательно, Δ G = L×ΔT/Гкр. Из этого выражения видно, что с ростом переохлаждения движущая сила кристаллизации увеличивается.
Следует отметить, что при кристаллизации возникает новая фаза, имеющая физическую поверхность раздела с жидкостью. Поэтому кристаллизация связана с затратами энергии на образование и рост данной поверхности раздела фаз. Величина этой энергии равна Es = sк-ж×S, где sк-ж — межфазная энергия на границе раздела фаз; S — поверхность растущего кристалла. В результате образования кристалла объемом V с поверхностью S энергия системы уменьшится на величину Δ Gv и увеличится на величину Es. Поэтому изменение суммарной энергии Гиббса будет равно
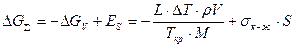 ,
,
где r и М— плотность твердого металла и его молярная масса. Анализ характера зависимости Δ Gå от объема кристалла V показывает, что она имеет экстремальный характер. Характер зависимостиΔ Gå от радиуса зародыша показан на рис. 2.2.
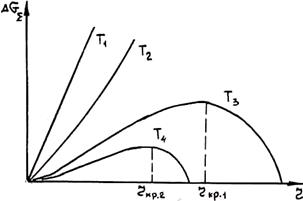
Кис. 2.2. Зависимость Δ Gå от радиуса зародыша r при разных температурах (T1>T2>Tкр>Т3>Т4)
В курсе металловедения получено следующее выражение для критического размера сферического зародыша, обеспечивающего максимум Δ Gå (читателю предлагается вывести его самостоятельно):
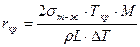 .
.
Из рис. 2.2 видно, что с понижением температуры, т. е. с увеличением переохлаждения, радиус критического зародыша становится меньше (rкр2 < rкр1). При этом уменьшается и энергия образования критического зародыша.
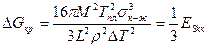 ,
,
где Esкр — поверхностная энергия зародыша критических размеров.
Пусть по каким-либо причинам в расплаве при данном переохлаждении возник зародыш радиуса r < rкр. Рост такого зародыша (увеличение r), как видно из рис. 2.2, сопровождается ростом Δ Gå. Поэтому данный зародыш не способен к росту и будет расплавляться. Если радиус зародыша r ³ rкр, то при присоединении к нему даже одного атома, т. е. с ростом его размеров, Δ Gå будет убывать. Поэтому данный зародыш сможет расти.
Центром кристаллизации будет называться зародыш, обязательно способный к росту. Критический зародыш (r = rкр) еще не является центром кристаллизации: он может потерять хотя бы одну частицу, т. е. стать дозародышем (r < rкр) и исчезнуть, или
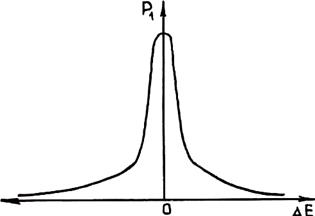
Рис. 2.3. Зависимость вероятности Р1 флуктуации энергии Е от величины флуктуационного отклонения Δ Е
присоединить хотя бы одну частицу (r > rкр) и получить возможность для роста, т. е. стать центром кристаллизации. Поэтому вероятность образования центра кристаллизации равна произведению вероятностей образования зародыша критических размеров p1 и присоединения к нему частицы р2, т. е. p = p1× p2.
Вероятность образования центра кристаллизации равна отношению числа возникших в единицу времени в единице объема расплава центров кристаллизации п к общему числу частиц в расплаве N, т. е. p = n/N. Величина п, называемая скоростью возникновения центров кристаллизации (СВЦК), является важнейшей кристаллизационной характеристикой металлического расплава. Очевидно, n = p× N = p1× p2× N. Размерность [п] — м-3× c-1.
Для того чтобы определить вероятность p1, рассмотрим вопрос о причинах появления зародышей в условиях возрастания суммарной энергии Гиббса Δ Gå. С точки зрения статистической физики характерные для равновесия значения термодинамических величин представляют собой лишь наиболее их вероятные при данных условиях значения. Практически отдельные микрообъемы жидкости в течение некоторых малых промежутков времени могут обладать значениями термодинамических величин, отличными от отвечающих данному равновесному состоянию. При этом большую часть времени система обладает равновесными термодинамическими характеристиками. Случайные отклонения какой-либо величины от ее равновесного значения называются флуктуациями. Вероятность обнаружения флуктуации зависит от ее величины. Чем больше флуктуация, т. е. отклонение от соответствующего равновесного значения, тем она менее вероятна и тем меньшее время система обладает соответствующим отличным от равновесного значением той или иной величины. Следует отметить, что кривая зависимости вероятности флуктуации от ее величины отличается острым максимумом в области нулевых флуктуаций (рис. 2.3). Поэтому с увеличением флуктуации вероятность ее появления резко снижается до нуля.
Благодаря наличию флуктуации энергии в расплаве будет находиться то или иное количество микрообъемов, обладающее избытком энергии, необходимым для совершения работы образования зародыша критических размеров Δ Gкр. Вероятность такой энергетической флуктуации равна pl = nк/N = exp(— Δ Gкр/kТ), где nк — число зародышей критического размера; N— общее число частиц в единице объема. Вероятность p1 и будет вероятностью возникновения зародыша критического размера в единице объема в единицу времени. Очевидно, что, чем больше Δ Gкр, тем большая требуется энергетическая флуктуация и меньше вероятность р1. Так как с увеличением переохлаждения Δ T Δ Gкр уменьшается, то с ростом Δ Т повышается р1. Вероятность присоединения к зародышу хотя бы одной частицы равна p2 = nsen× exp(—U/kT), где n s— число атомов в жидкости, находящихся в контакте с поверхностью зародыша; e = l/6 — вероятность скачка атомов в данном направлении; n — частота колебаний атомов в жидкости (n = 1013•c-l); U— энергия активации диффузии атомов в жидкости, определяющая подвижность атомов.
Таким образом, вероятность образования центра кристаллизации равна
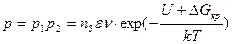 . (2.1)
. (2.1)
С учетом формулы (2.1) для вычисления скорости возникновения центров кристаллизации можно записать выражение
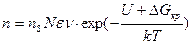 . (2.2)
. (2.2)
С увеличением переохлаждения Δ Gкр уменьшается, а U возрастает (снижается подвижность атомов). Поэтому зависимость п от Δ Т носит экстремальный характер (рис. 2.4). Подобную кривую получил впервые в начале века Г. А. Тамман при кристаллизации органических прозрачных жидкостей. Для чистых гомогенных металлических расплавов зависимость п от Δ T показана на рис. 2.5. Видно, что при переохлаждениях, меньших величины Δ Т1, называемой интервалом метастабильности по зарождению, зарождение центров кристаллизации практически не происходит. Для металлов ниспадающая ветвь кривой (см. рис. 2.4) не реализуется, так как подвижность атомов в металлах очень велика.
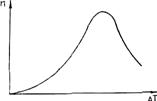
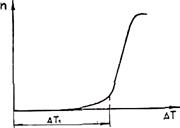
Рис. 2.4. Зависимость СВЦК " от переохлаждения ΔТ Рис. 2.5. Зависимость СВЦК от переохлаждения
для очистки гомогенных металлических расплавов
Для чистых гомогенных металлических расплавов величина интервала метастабильности очень велика. Расчеты показывают, что Δ T1» 0,2 Ткр. Реально подобная переохлаждаемость расплавов не наблюдается, так как в расплаве всегда находится достаточное количество примесей, инициирующих зародышеобразование.
Обычно самопроизвольную гомогенную кристаллизацию изучают на мельчайших каплях чистого металла. При этом считается, что при измельчении металла вероятность нахождения примеси в капле уменьшается. Зависимость n от Δ T для металлов можно аппроксимировать следующей степенной зависимостью: n = n0(ΔT — ΔT1)s, где показатель степени s находится в интервале от 1 до 3.
Выше речь шла о самопроизвольном зарождении кристаллов при затвердевании однокомпонентных расплавов (чистых металлов). Большинство литейных расплавов являются сплавами, т. е. высокотемпературными жидкими растворами. При этом продуктами кристаллизации чаще всего являются твердые растворы. При охлаждении до температуры ликвидуса жидкий сплав оказывается насыщенным по отношению к легирующему компоненту. Поэтому при дальнейшем снижении температуры наступает пересыщение и из расплава должны выделяться твердые кристаллы.
Свойства, не зависящие от количества вещества, называются интенсивными. К ним относятся температура и давление. Применительно к растворам Г. Н. Льюис ввел еще одну интенсивную величину, называемую парциально-молярной величиной компонента. Она представляет собой частную производную от какого-либо экстенсивного свойства W по числу молей этого компонента при постоянных давлении, температуре и числе молей остальных компонентов в растворе, т. е. qi = (дW/дni}p,T,n j¹i Например, парциально-молярная энергия Гиббса для (i -го компонента равна Gi = (дG/дпi)р,T, j¹i. Легко понять, что парциально-молярные свойства чистых веществ совпадают со значением этих свойств одного моля
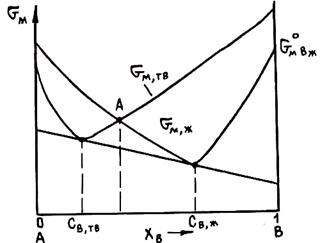
Рис. 2.6. Схема графического определения состава фаз, находящихся в равновесии при заданной температуре
вещества для заданных условий (G0A и G0B). Действительно, для вещества A G = nG0A и дG/дп = G0A. Величина дG/дпi называется химическим потенциалом mi, т. е. mI = (дG/дпi)P,T, j¹i Химический потенциал характеризует тенденцию вещества покинуть данную фазу и перейти в другую. Физически химический потенциал равен работе, которую нужно совершить, присоединяя к раствору одну частицу данного компонента. Интенсивные величины Т, Р и mI характеризуют условия термического, механического и химического равновесия. Для равновесия необходимо, чтобы во всех фазах были одинаковые температуры, давления и химические потенциалы компонентов.
Приравняв величины mI для каждого компонента во всех фазах, получим систему уравнений, которая совместно с уравнениями материального баланса позволяет найти составы находящихся в равновесии фаз. Для двойных сплавов этот подход хорошо реализуется графически. На рис. 2.6 показан пример графического метода определения состава твердого раствора и находящейся с ним в равновесии при некоторой температуре Т жидкой фазы. При равновесии должны выполняться условия mтвА = mжА и mтвВ = mжВ, а если mВ = дG/дпв, то составы фаз должны обеспечивать выполнение равенства
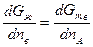 или
или 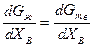
Геометрически это означает, что составы определяются общей касательной к кривым зависимости энергий Гиббса твердой и жидкой фаз при данной температуре. Эти равновесные составы GтвВ и GжВ показаны на рис. 2.6. Иногда считают, что искомые составы отвечают точкам пересечения кривых мольных энергий фаз, например точке А, т. е. точкам, где равны энергии фаз. Однако равновесные фазы обеспечивают минимум суммарной энергии обеих фаз, а он достигается только при mжА = mтвА и mжВ = mтвВ.
Таким образом, при достижении пересыщения из раствора могут выделяться твердые растворы, концентрации которых отличаются от исходной. При этом для начала процесса кристаллизации необходимо пересыщение раствора, достигаемое его переохлаждением относительно температуры ликвидуса.
Движущая сила, создающая условия для выделения твердой фазы в сплавах, определяется так же, как и для металлов, по изменению энергии Гиббса системы.
При кристаллизации сплавов вместо справедливого для однокомпонентного металла выражения Δ Gм = L× Δ T'/Tпл следует применять выражение Δ Gж-a С учетом этого для критического размера зародыша получим выражение rкр = 2sМ/(r× Δ Gмж-а). Работа образования критического сферического зародыша равна 16 pМ2s3/[ З r2( Δ Gж-а)2].
Следует отметить, что зарождение центров кристаллизации сплавов, как показали исследования, качественно описывается той же зависимостью от переохлаждения, что и у металлов. При этом переохлаждение отсчитывается от температуры ликвидуса. Величину Δ Gмж-a можно также вычислить по формуле L Δ T/Тл, где L — теплота кристаллизации, выделившаяся при кристаллизации единицы массы сплава при данной температуре.
Для образования зародышей твердых растворов необходимы не только рассмотренные выше энергетические флуктуации, но и флуктуации концентрации. Концентрационные флуктуации представляют собой участки растворов, состав которых отличается от среднего. Например, в жидком чугуне при средней концентрации углерода 3,4 % обнаруживаются участки размером 10-8 см, состоящие из одного углерода. По-видимому, именно эти участки являются основой возникновения зародышей графита при кристаллизации чугуна.
Рассмотренный выше процесс кристаллизации сплавов называется диффузионным, так как при кристаллизации в этих случаях происходит перераспределение компонентов между фазами путем диффузии.
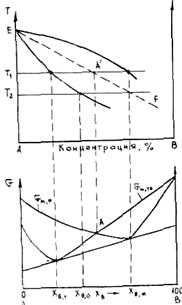
Рис. 2.7. Схема бездиффузионной кристаллизации
Возможен бездиффузионный процесс кристаллизации сплавов, когда состав выделяющейся твердой фазы строго совпадает с составом исходного жидкого сплава. На рис. 2.7 представлена диаграмма состояния и зависимость G от состава для жидкого сплава и сплава в твердом состоянии. Зависимость G приведена при T = T1. Точка A` отвечает равенству мольных энергий Гиббса твердой и жидкой фаз. На диаграмме состояния ей соответствует точка А`. Если аналогичные точки перенести на диаграмму при других температурах, то получим линию EF, отвечающую равенству Gт = Gж. Если сплав резко переохладить ниже этой линии (например до T = T2), то Gт станет меньше Gж и произойдет бездиффузионная кристаллизация с выделением твердой фазы концентрации С0. Анализ показывает, что для этого нужны очень большие переохлаждения (порядка нескольких десятков и даже сотен.градусов) В обычной литейной практике они не достигаются, так как требуют очень больших скоростей охлаждения при малых объемах сплава.
ГЛАВА 2.2. ТЕОРИЯ ГЕТЕРОГЕННОГО ЗАРОДЫШЕОБРАЗОВАНИЯ
Большинство металлов затвердевает при значительно меньшем переохлаждении, чем максимальное переохлаждение Δ Tmах = 0,2Tкр предсказываемое теорией гомогенного образования зародышей. Это обусловлено наличием в расплаве инородных твердых частиц которые облегчают возникновение зародышей, Рассмотрим простейший случай образования зародыша на плоской поверхности включения.
Пусть зародыш ограничен частью сферической поверхности радиуса R (рис. 2.8). Изменение полной мольной энергии сплава при образовании зародыша в данном случае имеет вид
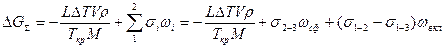 ,
,
где s2-3 и s1-2 — межфазные энергии на границах сплав — зародыш и зародыш — включение; wсф и wвкл — площади поверхностей зародыша на границе со сплавом и на границе с включением; s1-3 — межфазная энергия на границе между включением и сплавом. Выполнив геометрические преобразования (предлагаем читателю выполнить их самостоятельно), получаем следующие формулы для вычисления V, wсф и wвкл:
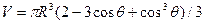 ;
;
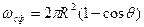 ;
; 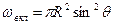 .
.
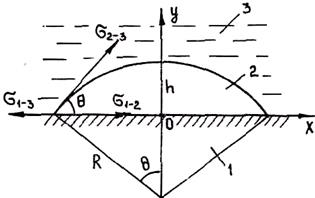
Рис. 2.8. Схема зарождения кристалла на плоской поверхности включения:
/ — включение; 2 — зародыш; 3 — сплав
Подставив эти выражения в формулу для Δ Gå и выполнив исследования на максимум, получаем следующие выражения для вычисления энергии образования критического зародыша на включении и соответствующего ему размера Rкр:
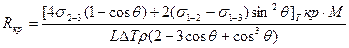 ;
;
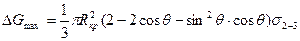 .
.
Предлагаем читателю получить эти формулы самостоятельно.
При полном несмачивании включения сплавом (q = p) имеем сферический зародыш. При этом Δ Gmах = 4p(R*)2/3s2-3. При полном смачивании (q = 0) ΔGmах = 0. Сравним работу образования зародыша на включении при 0 < q < 180° с работой гомогенного образования сферического зародыша:
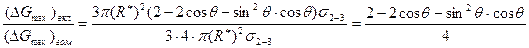 <1 при 0 < q < p.
<1 при 0 < q < p.
Таким образом, работа образования критического зародыша на включении меньше, чем в гомогенном расплаве. СВЦК при этом вычисляется по ранее приведенным формулам, если вместо Δ Gmах в них подставлять полученное выше выражение для расчета максимального изменения энергии Гиббса при образовании зародыша на включении. Так как Δ Gmах при этом меньше, чем при гомогенном зарождении, то СВЦК при наличии включений увеличивается. Очевидно, что Δ Gmах становится меньше с уменьшением q, т. е. с усилением смачиваемости включения сплавом. Поэтому СВЦК увеличивается с уменьшением q.
Следует отметить, что не любые твердые включения резко увеличивают СВЦК. Из приведенных выражений видно, что это произойдет, если малы значения q и s2-3. Очевидно, что s2-3 будет тем меньше, чем ближе кристаллическая структура включения к кристаллической структуре зародыша. По правилу ориентационно-размерного соответствия, предложенного П. Д. Данковым и С. Т. Конобеевским, необходимо, чтобы включения имели один тип кристаллической решетки со сплавом. При этом параметры решеток не должны сильно отличаться. Такие включения называют изоморфными. Указанное правило было обосновано П.Д. Данковым следующим образом. На подкладке может образоваться ориентированный относительно ее решетки зародыш (он является как бы продолжением решетки включения) или произвольно ориентированный зародыш (как при гомогенной кристаллизации). Если параметры решеток различаются, то решетка зародыша, взаимодействуя с атомами решетки включения, упруго деформируется (расширяется или сжимается со сдвигом).
Если работа деформации меньше энергии образования трехмерного неориентированного зародыша, то зародышеобразования на включении не произойдет. Выполнив соответствующие расчеты для хлористого натрия, П. Д. Данков нашел, что ориентированное зарождение прекращается при различии в параметрах решеток соли и включения, равном 9 %. Практическая проверка формулы Данкова показала, что она дает завышение предельно допустимого отклонения параметров решеток. Практически при кристаллизации соли хлористого натрия на галените ориентированное зарождение прекращалось при Δ а/а = 5,7 %. В качестве примеров ориентированного зарождения центров кристаллизации металлических расплавов можно привести рост медных кристаллов (а = 3,6Å) на никелевых включениях (а = 3,52Å), кристаллов a-латуни (а = 3,68Å) на серебре (а = 4,08Å). Измельчение кристаллического зерна в отливках вследствие увеличения СВЦК путем введения в расплав изоморфных
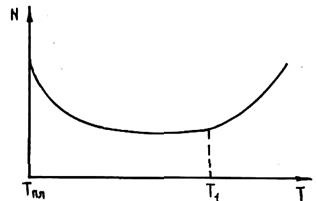
Рис. 2.9. Зависимость числа кристаллических зерен на единице площади шлифа от перегрева металла
примесей широко применяется на практике и называется модифицированием первого рода. Например, при введении в жидкий алюминий титана или циркония образуются тугоплавкие интерметаллические соединения TiAl3 и ZrА13, имеющие изоморфную с алюминием кристаллическую решетку. Достаточно ввести в сплав десятые доли процента титана или циркония, как размер кристаллического зерна уменьшается в десятки и сотни раз.
Затравочное действие твердых включений зависит от термовременной обработки расплава. На рис. 2.9 приведена зависимость числа кристаллических зерен N на единицу площади шлифа от температуры обработки жидкого сплава. Видно, что с ростом температуры перегрева сплава N уменьшается. Начиная с некоторой, хотя и очень высокой, температуры, ниспадающая кривая переходит в восходящую. Увеличение СВЦК при высоких перегревах объясняется тем, что при этом дезактивированы все включения и сплав кристаллизуется по гомогенному механизму в условиях очень больших переохлаждении.
Очевидно, что если в расплаве содержится большое число истинно изоморфных примесей, то их дезактивация при перегреве не происходит.
При увеличении перегрева сплава кроме отмеченных выше явлений может происходить растворение включений в расплаве или, наоборот, выделение их из расплава. В зависимости от характера выделяющихся или растворяющихся включений изменяется кристаллическая структура сплава. Например, при перегреве магниевых сплавов, легированных алюминием, марганцем и железом, при некоторой температуре происходит растворение алюминиево-железистых соединений в расплаве. При охлаждении сплава в процессе кристаллизации эти соединения выделяются из расплава в виде компактных изоморфных частиц, что увеличивает СВЦК и измельчает структуру. Этот прием широко применяется в практике магниевого литья и носит название модифицирования перегревом. При перегреве чугуна происходит полное растворение углерода, что по аналогичным перегреву магниевых сплавов причинам приводит к резкому увеличению дисперсности выделяющихся при кристаллизации чугуна включений графита.
В заключение отметим, что при кристаллизации на примесях сохраняется характер изменения СВЦК с уменьшением переохлаждения. Однако при этом резко сокращается интервал метастабильности и повышается количественный уровень СВЦК. Это объясняется не только непосредственным влиянием переохлаждения на работу образования и размер критического зародыша, но и увеличением числа неизоморфных включений, удовлетворяющих сформулированному выше условию ориентированного роста.
Действительно, по данным Д. Тернбала и Б. Воннегута, энергия упругого деформирования кристалла на подкладке может
быть оценена по формуле Δ G = (авкл + aм)2/a2м, а движущая процесс разность энергий Гиббса при увеличении переохлаждения возрастает. Поэтому при увеличении переохлаждения все большее число включений удовлетворяет условию ориентированного зарождения.
ГЛАВА 2.3. МЕХАНИЗМ И КИНЕТИКА РОСТА КРИСТАЛЛИЧЕСКИХ ЗАРОДЫШЕЙ
Образовавшиеся центры кристаллизации самопроизвольно увеличивают свои размеры. Кристалл растет путем присоединения к нему атомов из окружающего расплава. Способ присоединения атомов к поверхности растущего зародыша определяется строением поверхности раздела фаз, распределением и количеством дефектов на этой поверхности. В соответствии с современной теорией кристаллизации механизм роста кристаллов определяется состоянием поверхности их граней. В зависимости от этого фактора можно выделить следующие механизмы роста кристаллов.
1. Рост кристаллов на атомно-гладких поверхностях кристалла. В этом случае все атомы поверхности раздела кристалл — расплав расположены на одной плоскости, на которой нет образований адсорбированных атомов типа уступов, ступеней и т. п. Образование уступов, ступеней, выступов из групп атомов называется процессом огранки поверхности, которая при этом называется ограненной.
При атомно-гладкой поверхности зародыша (рис. 2.10, а) возможен только послойный рост кристалла путем образования на гранях двумерных зародышей, которые разрастаются
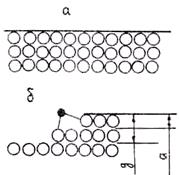
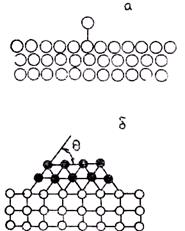
Рис. 2.10. Типы морфологии поверхности растущего Рис. 2.11. Рост кристалла путем присоединения
кристалла: а — атомно-гладкая поверхность; отдельных атомов (а) и двумерных зародышей (б)
б — ограненная поверхность
затем вдоль этих граней. Рост путем присоединения отдельных атомов в данном случае энергетически неустойчив, так как каждый адсорбированный поверхностью атом имеет только одного ближайшего «соседа» и, следовательно, только одну связь (рис. 2.11, а). В таких условиях он не может устойчиво закрепиться на поверхности. Поэтому рост может происходить только путем образования и закрепления тонких, одно-, двух- и трехатомных по толщине, плоских образований атомов, называемых двумерными зародышами.
На рис. 2.12 приведена схема двумерного зародыша на грани растущего кристалла. При образовании такого зародыша поверхность кристалла увеличивается на Δ S = 4×2x×d = 8x×d, т. е. на величину поверхности четырех боковых граней. Соответствующее возрастание поверхностной энергии равно As = 8xds = 8×xs', где s' = `ds — поверхностная энергия, приходящаяся на единицу периметра зародыша толщиной d. Если энергию Гиббса зародыша с основанием 1 см2 обозначить j2, а энергию Гиббса, приходящуюся на то же число атомов жидкости, — j1, то для полного изменения энергии при зародышеобразовании можно написать следующее выражение:
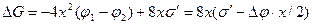 .
.
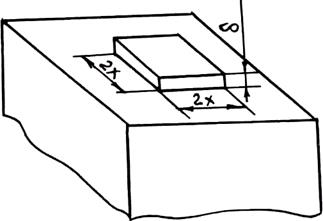
Рис. 2.12. Схема образования двумерного зародыша на грани кристалла
Подставив в это выражение Δ j = j1 — j2 = Δ H`× Δ T/Ткр (см. гл. 2.1), где Δ H' — теплота плавления зародыша с основанием 1 см2, получаем
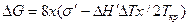 .
.
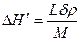 ;
; 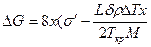 .
.
Исследуем эту зависимость на экстремум:
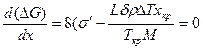 .
.
Из этого уравнения находим выражение для вычисления критического размера зародыша хкр, при котором ΔG достигает максимума:
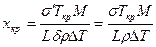 .
.
Работа образования этого зародыша вычисляется по формуле
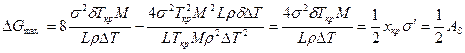
Сравнение полученных формул для вычисления размера и работы образования двумерного критического зародыша с соответствующими выражениями для трехмерного зародыша (см. гл. 2.1) показывает, что критический размер двумерного зародыша в 2 раза меньше, чем трехмерного, меньше и работа образования критического двумерного зародыша. Вероятность образования в единице объема двумерного зародыша равна
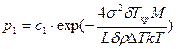 .
.
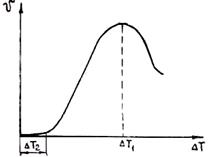
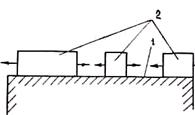
Рис. 2.13. Зависимость линейной скорости Рис. 2.14. Схема одновременного роста нескольких
роста кристалла от переохлаждения двумерных зародышей:
/—грань кристалла; 2— зародыши
Вероятность присоединения к зародышу в единицу времени хотя бы одного атома равна р2 = с2×ехр(—U/kT). Вероятность образования центра кристаллизации вычисляется по формуле
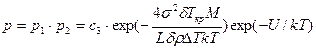 ,
,
где с3 = с1×с2 — некоторая постоянная для данного вещества величина.
Скорость роста кристалла пропорциональна вероятности р, т. е.
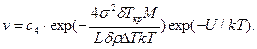
Характер зависимости линейной скорости роста от переохлаждения Δ Т показан на рис. 2.13.
Величина интервала метастабильности при росте кристаллов Δ Т2 значительно меньше, чем Δ Т1 при зарождении. При этом, как правило, реализуется только восходящая ветвь кривой (см. рис. 2.13). Зависимость можно аппроксимировать следующей функцией:
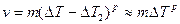 ,
,
где показатель степени 1 £ р £ 2. Двумерный зародыш быстро растет, последовательно присоединяя к своим боковым граням ряды атомов так, что слой, образованный зародышем, покрывает грань кристалла. После заполнения грани наступает пауза, длительность которой t ~ ехр(4s`хкp/kT) обратно пропорциональна вероятности р1. Затем наращивается новый слой и т. д. У кубического кристалла в идеальном случае все шесть граней перемещаются с одинаковой скоростью в направлениях своих нормалей.
Когда грань кристалла достаточно велика, длительность ее заполнения t значительно больше времени ожидания появления нового зародыша t. В этом случае возможны образование и независимый рост двух или нескольких зародышей (рис. 2.14).
2. На атомно-шероховатых поверхностях кристаллов могут надежно адсорбироваться отдельные атомы жидкости (рис. 2.10, б). При этом происходит так называемый нормальный рост кристаллов, когда атомы расплава беспорядочно присоединяются к любым точкам поверхности. В этом случае грань кристалла перемещается однородно по нормали к самой себе. Соответствующий расчет показывает, что линейная скорость роста в данном случае пропорциональна переохлаждению:
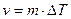 ,
,
где m = n×L×M×exp(—U/kT)/(T2nep×R); R — универсальная газовая постоянная; n — частота колебаний атомов в жидкости.
3. Механизм роста существенно изменяется, если на поверхности растущего кристалла имеются какие-нибудь выступы, которые облегчают закрепление частиц жидкости на поверхности кристалла. Очень часто такие выступы образуются в местах выхода винтовых дислокаций. Кристаллы, на гранях которых имеются ступеньки, возникшие в результате выхода винтовых дислокаций, растут послойно путем присоединения атомов расплава к этим ступенькам. Не приводя вывода, запишем формулу для вычисления линейной скорости в этом случае:
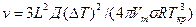 ,
,
где s — межфазное натяжение на границе кристалл — жидкость; Vm — молярный объем жидкости; Д — коэффициент диффузии.
Из формулы видно, что линейная скорость роста v пропорциональна (ΔT)2. Дислокационный послойный рост происходит при значительно меньших переохлаждениях, чем рост двумерных зародышей. Интервал метастабильности по росту в этом случае равен нулю.
На рис. 2.15 приведены результаты исследования механизма роста в зависимости от величины переохлаждения на фронте кристаллизации. Видно, что при малых переохлаждениях рост осуществляется путем образования двумерных зародышей, при больших переохлаждениях
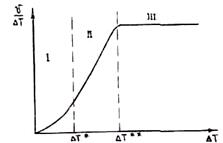
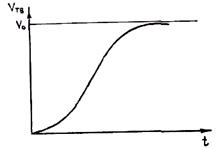
Рис. 2.15. Зависимость механизма роста кристаллов от Рис. 2.16. Кинетическая кривая криcталлизации
величины переохлаждения на фронте кристаллизации:
I — зона образования двумерных зародышей;
II — промежуточная зона;
III — зона нормального роста кристаллов
— по нормальному механизму. При этом фронт не огранен, т. е. имеет вид атомно-шероховатой поверхности. В промежуточной зоне переохлаждения рост может осуществляться по дислокационному механизму. В реальных условиях литья, как правило, реализуется нормальный механизм роста.
ГЛАВА 2.4. КИНЕТИКА КРИСТАЛЛИЗАЦИИ. ФОРМУЛА КОЛМОГОРОВА
Рассмотренные выше характеристики процесса кристаллизации—скорость возникновения центров кристаллизации п и линейная скорость роста кристаллов v — являются важнейшими кристаллизационными характеристиками, определяющими кинетику кристаллизации. Математической величиной, описывающей кинетику кристаллизации, является временная зависимость объема закристаллизовавшейся жидкости.
Решение задачи о кинетике нарастания твердой фазы при кристаллизации было впервые выполнено А. Н. Колмогоровым. Используя аппарат теории вероятности, можно получить следующую формулу:
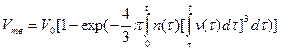 ,
,
где n(t) = v(т) — скорость возникновения центров кристаллизации и линейная скорость их роста как функции времени; V0 — начальный объем расплава. Формула справедлива для сферического зародыша. Если принять в ней n = const и v = const, то после интегрирования нетрудно получить формулу Колмогорова
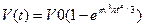 . (2.3)
. (2.3)
Графически она описывается кривой, показанной на рис. 2.16. Видно, что на начальных и конечных временных участках наблюдается незначительный темп нарастания твердой фазы. Замедление кристаллизации в конце объясняется тем, что растущие кристаллы начинают соприкасаться друг с другом при малом и непрерывно уменьшающемся количестве жидкости. Так как в конце кристаллизации объем жидкости мал, то основное положение вывода V0 >> Vкр не соблюдается. Поэтому из формулы вытекает неправдоподобный результат, что продолжительность полного затвердевания объема равна бесконечности. При этом погрешность формулы тем больше, чем меньше кристаллов и чем больше размер зерна. Принято считать, что формула обеспечивает расчет с погрешностью не более 1 %, если выполняется условие V0(n/v)3/4 > 200. Анализ показывает, что это условие, как правило, соблюдается при затвердевании реальных сплавов.
В обобщенном виде уравнение кинетики кристаллизации можно записать следующим образом:
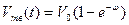 ,
,
где 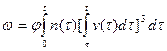 ; (j — коэффициент, учитывающий конфигурацию кристаллита (j = 4p/3 для шара и j = 8 для кубических кристаллитов).
; (j — коэффициент, учитывающий конфигурацию кристаллита (j = 4p/3 для шара и j = 8 для кубических кристаллитов).
Рассмотрим кинетику кристаллизации при объемном затвердевании. Возьмем некоторый объем сплава Vo, который охлаждается при малой интенсивности теплообмена с внешней средой. В этих условиях можно пренебречь перепадом температур по объему и рассматривать, что кристаллизация происходит путем равномерного выделения по всему объему твердой фазы. Такое затвердевание называют чисто объемным. Пусть при t = 0 температура расплава равна T = Tкр— Δ T1, где Δ T1 — интервал метастабильности по зарождению кристаллов. Напишем уравнение теплового баланса:
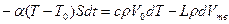 ,
,
где a — коэффициент теплоотдачи с наружной поверхности объема; S — поверхность охлаждения; T0 —температура среды; L, с и р — удельная теплота кристаллизации, теплоемкость и плотность расплава; Vтв — объем металла, затвердевшего за время t; dT— изменение температуры за время dt. Для оценки зависимости параметров п и v от переохлаждения примем v = m Δ T = m(Tкр – T), n = n0( Δ T – Δ T1)2 = n0(Tкp – T – Δ Tl)2 приΔ T >Δ T1 и n = 0, если Δ T £ Δ T1. Проинтегрируем это уравнение при начальном условии: при t = 0 Vтв = 0 и T = Tкр – Δ T1. Кроме того, в левой части уравнения примем T = Ткр ввиду малой величины переохлаждения Δ T при кристаллизации металлов.
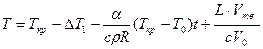 ,
,
Подставив в это уравнение выражение для Vтв (см. формулу Колмогорова), получаем интегральное уравнение, описывающее изменение температуры металла при его объемной кристаллизации:
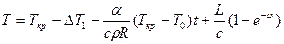 . (2.4)
. (2.4) 
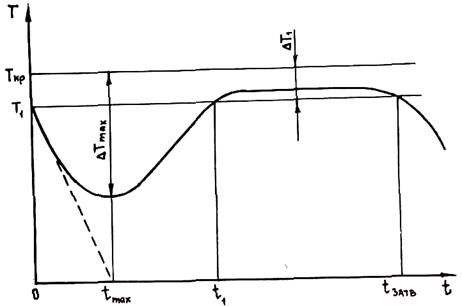
Рис. 2.17. Изменение температуры расплава при его объемной кристаллизации
Решить это уравнение возможно только с применением численных методов на ЭВМ. Изменение температуры во времени показано на рис. 2.17 (расчетные данные). Подобный характер температурной кривой подтверждается многочисленными экспериментами.
Если бы в расплаве не выделялась теплота кристаллизации, то температура уменьшалась бы в соответствии со штриховой линией на рис. 2.17, которая отвечает уравнению T = Tкр – Δ T1 - (Ткр – Т0)×t×a /(c×r×R). Однако выделяющаяся теплота кристаллизации замедляет темп охлаждения, и в момент t = tmax dT/dt = 0, что соответствует минимуму температуры, или максимуму переохлаждения ( Δ T = Tкр – Т). При t > tmax dT/dt > 0, а температура сплава повышается, пока переохлаждение не станет меньше интервала метастабильности по зарождению Δ T1. При t > t1 Δ T < Δ Т1 и зарождение новых кристаллов в расплаве прекращается. Далее будет происходить только рост образовавшихся в период 0 < t < t1 центров до полного затвердевания металла при t = tзатв. Этот подъем температуры называется рекалесценцией.
Приближенное решение уравнения, выполненное с использованием метода итераций, позволило получить следующие формулы для расчета параметров температурной кривой:
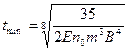 ;
;
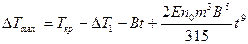 ;
;
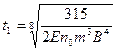 ;
; 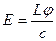 ;
; 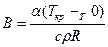 .
.
Для вычисления количества центров кристаллизации, зародившихся на первом этапе кристаллизации, получена следующая приближенная формула:
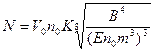 ,
,
где К» 3,5.
Расчеты на ЭВМ и эксперименты показали достаточную точность этих формул.
Величина N фактически определяет число кристаллических зерен в затвердевшей отливке, так как при t > t1 новые зерна не зарождаются. Приняв, что V0 = Npd3/6, где d — эффективный диаметр зерна, с учетом формулы для вычисления N легко найти следующее выражение для оценки величины кристаллического зерна при объемной кристаллизации:
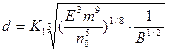 ,
,
где K1 = 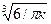 .
.
Из полученной формулы видно, что для измельчения зерна необходимо увеличить СВЦК. n0 и уменьшить линейную скорость роста т. Видно, что зерно измельчается при повышении коэффициента теплоотдачи а и снижении удельной теплоты кристаллизации L.
Рассмотренная выше схема чисто объемной кристаллизации, когда твердая фаза растет от отдельных центров во всем объеме жидкости, может быть реализована для чистых металлов при малом значении критерия Био (Bi = aR/l), т. е. при малых значениях a и R. При Bi > 0,l, как правило, объемная кристаллизация в центральных объемах сопровождается последовательным продвижением затвердевшей корки с поверхности охлаждения сплава к центру отливки. При объемной кристаллизации величина максимального переохлаждения Δ Tmах невелика (для алюминия не более 1—5°С, для железа — до 40 °С). Длительность первого этапа (t £ t1) составляет относительно небольшую долю от общего времени затвердевания. Как правило, мы измеряем термопарой температуру сплава на втором этапе затвердевания, когда переохлаждение (Δ T < Δ T1) очень мало. При этом температура, фиксируемая термопарой, близка к Ткр.
Изложенная теория показывает, какое большое значение для управления формированием кристаллического строения отливки кроме теплового режима имеет целенаправленное воздействие на кристаллизационные параметры п и v.
ГЛАВА 2.5. ПЕРЕРАСПРЕДЕЛЕНИЕ ПРИМЕСЕЙ НА ФРОНТЕ КРИСТАЛЛИЗАЦИИ СПЛАВОВ. ВНУТРИКРИСТАЛЛИЧЕСКАЯ ЛИКВАЦИЯ
При кристаллизации сплавов концентрации компонентов в фазах, находящихся в равновесии, как правило, различаются. Отношение концентраций компонента в находящихся в равновесии твердой и жидкой фазах k0 = CS/CL. называется равновесным коэффициентом распределения. На рис. 2.18, а приведен участок диаграммы состояния сплава, температура ликвидуса которого уменьшается с увеличением концентрации легирующего компонента В. Очевидно, что в этом случае коэффициент распределения k0 < 1 (обычно 0,2 £ k0 £ 0,6).
При повышении температуры ликвидуса (рис. 2.18, б) коэффициент распределения k0 > 1. Обычно в этом случае k0 < 3. Однако, например, в системе Ge — В k0 достигает 15.
Дальнейшее изучение кристаллизации будем проводить при k0 < 1, так как эта величина является характерной для большинства литейных сплавов.
Рассмотрим несколько частных случаев.
1. Скорость роста кристалла настолько мала, что диффузия успевает выравнять концентрации вещества по объему твердой и жидкой фаз. При этом концентрации будут соответствовать равновесным значениям, определенным для данной температуры по диаграмме состояния. На рис. 2.19 показано распределение концентрации по длине твердой и жидкой фаз. Ось ординат, которая
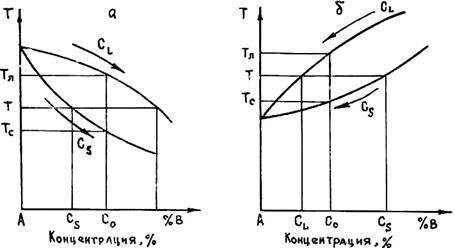
Рис. 2.18. Диаграммы состояния сплавов с понижением (а) и повышением (б) температуры ликвидуса в зависимости от роста концентрации легирующего компонента
соответствует концентрации, является подвижной и совпадает с положением фронта кристаллизации. В этом случае k0 = СS/СL.
2. Ситуация, более приближенная к реальной, имеет место, когда в твердой фазе при данной скорости роста кристалла не происходит диффузионного перераспределения вещества (Дтв = 0). Однако в жидкости, вследствие интенсивного перемешивания расплава, концентрации выравниваются. Так как Дтв = 0, то состав твердой фазы, образовавшийся на предыдущих стадиях, остается неизменным. Так как при k0 < 1 по мере кристаллизации CS увеличивается (см. рис. 2.18, а), то концентрация вещества будет возрастать по длине твердой фазы, оставаясь на фронте кристаллизации равной соответствующему значению, определенному по диаграмме состояния. Распределение концентраций по длине фаз в рассматриваемом случае приведено на рис. 2.20. Так как концентрация жидкости cl больше концентрации твердой фазы на фронте кристаллизации CS, то при затвердевании жидкости на фронте кристаллизации выделяется легирующий компонент. Например, если за время dt затвердевает dm жидкости, то количество выделившегося вещества равно (Cl—CS)dm. В данном случае это вещество равномерно распределяется по объему жидкости.
Явление, связанное с неравномерным распределением концентрации вещества, называется ликвацией. В рассматриваемом случае наблюдается максимальная ликвация в твердой фазе, называемая внутрикристаллической ликвацией. При этом внутренние слои сферического
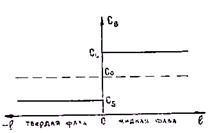
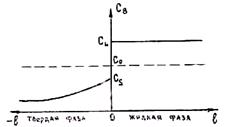
Рис. 2.19. Распределение концентрации по длине Рис. 2.20. Распределение концентраций по
жидкой и твердой фаз при малой скорости длине фаз при Дтв = 0 и Дж ® ¥
кристаллизации (k0 < 1)
кристаллита будут иметь меньшую концентрацию легирующего компонента, чем поверхностные (при слои сферического k0 < 1). Среднее содержание вещества в твердой фазе будет всегда меньше определенного по диаграмме состояния, а в жидкости, наоборот, средняя концентрация превысит равновесные значения. Это приведет к смещению влево линий солидуса на диаграмме состояния. Положение равновесной и неравновесной линии солидуса показано на рис. 2.21. Неравновесная линия солидуса отвечает средним значениям концентрации компонента в твердой фазе. Из рис. 2.21 видно, что при равновесной температуре солидуса Тсол. р часть жидкости остается незатвердевшей. Массовую долю этой жидкости можно найти по правилу рычага. Она равна отношению отрезков ae/ab. Полное затвердевание жидкости произойдет при более низкой температуре неравновесного солидуса Tсол.н.р, которая ниже равновесного значения на величину отрезка Δ T = ес.
Для выравнивания концентрации по объему кристаллитов применяют гомогенизационный отжиг, т. е. выдержку сплава при повышенных температурах, при которых значение коэффициента диффузии Дтв достаточно для диффузионного относительно быстрого выравнивания концентраций. Так как при перемешивании жидкости вещество, выделившееся на фронте затвердевания, отводится и равномерно распределяется по ее объему, то в этом случае k0 = СS/СL.
3. При переходе к более высоким скоростям роста, которые обычно имеют место, реализуется диффузионно контролируемый режим кристаллизации. Перенос вещества с фронта кристаллизации в объем жидкости осуществляется путем диффузии, и распределено оно в ней неравномерно. Примесь, выделяющаяся при образовании твердой фазы (k0 < 1), накапливается у движущегося фронта кристаллизации, и ее концентрация на фронте СL0 превышает концентрацию в объеме СL (рис. 2.22). В данном случае k0 = CS/CL0.
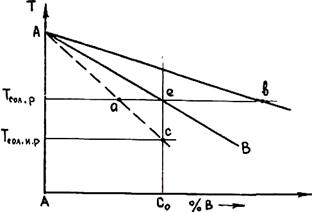
Рис. 2.21. Положение равновесной и неравновесной линий солидуса на диаграмме состояния сплава
В процессе перемещения фронта кристаллизации концентрация СL0 повышается, потому что не вся примесь отводится в расплав. Это приводит к увеличению концентрации растворенного вещества как в жидкости, так и в твердой фазе. При некотором положении фронта кристаллизации концентрация в твердой фазе достигает С0, а в расплаве на фронте — СL0 = С0/k0. Соответствующее этому моменту распределение концентраций показано на рис. 2.23.
В этих условиях можно написать следующее уравнение баланса вещества для фронта кристаллизации:
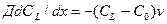 .
.
Здесь выражение в левой части определяет количество вещества, отведенного от фронта в расплав, а выражение в правой части — количество вещества, выделившегося при кристаллизации. Проинтегрировав это уравнение при начальных условиях x = 0, CL =C0/k, получаем выражение, описывающее распределение концентрации примеси в расплаве вблизи фронта кристаллизации:
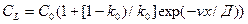 . (2.5)
. (2.5)
На некотором расстоянии х = d cl практически равно С0. Величина d называется толщиной диффузионного пограничного слоя.
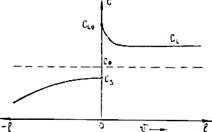
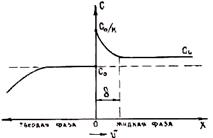
Рис. 2.22. Распределение примеси при диффузионно Рис. 2.23. Распределение концентраций в
контролируемой кристаллизации момент достижения их предельных значений
на фронте кристаллизации
Она зависит от интенсивности движения жидкости. С увеличением скорости движения жидкости, особенно при перемешивании, d быстро убывает. Если жидкость покоится, то d = Д/(v× k0).
ГЛАВА 2.6. КОНЦЕНТРАЦИОННОЕ ПЕРЕОХЛАЖДЕНИЕ. УСЛОВИЕ УСТОЙЧИВОСТИ ПЛОСКОГО ФРОНТА КРИСТАЛЛИЗАЦИИ СПЛАВОВ
При достаточно больших скоростях кристаллизации концентрация легирующего компонента перед фронтом кристаллизации распределена неравномерно. Распределение концентрации в расплаве описывает формула (2.5), из которой видно, что по мере удаления от фронта cl более или менее быстро убывает.
Как видно из диаграммы состояния (см. рис. 2.18, а), температура ликвидуса с ростом концентрации легирующего компонента уменьшается. Если принять линии ликвидуса и солидуса прямыми (рис. 2.24), то для зависимости температуры ликвидуса от концентрации компонента cl справедливо следующее уравнение:
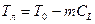 ,
,
где Т0 — температура плавления металла, являющегося основой сплава; m = tg a — тангенс угла наклона прямой ликвидуса к оси концентрации компонента В.
С учетом зависимости СL(х) получаем уравнение
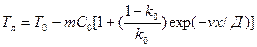 ,
,
из которого следует, что температура ликвидуса сплава перед фронтом кристаллизации по мере удаления от него увеличивается, достигая при х = d значения Tл, отвечающего исходной концентрации сплава С0. Температура ликвидуса сплава на фронте кристаллизации (СL0 = С0/k0) равна Tф = T0 – mC0/k0. Схема распределения температуры ликвидуса в расплаве перед фронтом кристаллизации приведена на рис. 2.25. При охлаждении сплава в нем возникает температурный градиент через твердую фазу, равный на фронте кристаллизации GR = (dT/dx)x=0. Примем линейное распределение температуры сплава Т (х) в пределах пограничного слоя d. При этом T(x) = Tф + GR×x. Следует отметить, что, приняв Т (х = 0) = Тф, мы пренебрегаем термическим переохлаждениемΔ T.
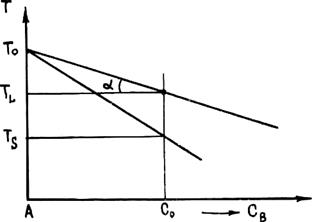
Рис. 2.24. Схема к выводу зависимости Тл(СL)
На рис. 2.25 линия 1 отвечает кривой изменения температуры ликвидуса, а линия 2 — распределению температуры сплава перед фронтом кристаллизации. В пределах слоя 0 £ х £ dп температура сплава ниже температуры ликвидуса, т. е. сплав является переохлажденным. Это переохлаждение обусловлено изменением состава сплава в процессе кристаллизации и называется концентрационным переохлаждением Δ Тк в отличие от рассмотренного в гл. 2.1 термического переохлаждения Δ T, обусловленного падением температуры сплава за счет теплоотвода ниже температуры ликвидуса на фронте кристаллизации или в какой-либо части объема жидкости.
Наличие концентрационного переохлаждения создает условия для нарушения плоского характера фронта кристаллизации. На фронте кристаллизации всегда имеются шероховатости с выступами микроскопических размеров. Вершины этих выступов, находящиеся перед фронтом кристаллизации, попадают в зону концентрационного переохлаждения, что существенно повышает скорость их продвижения в глубь расплава, так как с увеличением переохлаждения любой природы линейная скорость роста кристалла возрастает. Поэтому скорость продвижения в глубь расплава вершин выступов больше, чем оснований, находящихся на фронте, где ΔTк = 0 (см. рис. 2.25). В результате высота выступов увеличивается и плоский фронт кристаллизации нарушается.
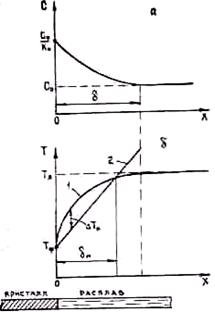
Рис. 2.25. Распределение концентрации (а) и температуры ликвидуса (б) перед фронтом кристаллизации
Таким образом, плоский фронт будет устойчивым, если отсутствует концентрационное переохлаждение. Очевидно, что если GR ³ (dTл/dx)x=o, то концентрационное переохлаждение отсутствует, так как при этом прямая распределения температур Т (х) касается кривой Тл(х) на фронте кристаллизации или лежит выше ее (рис. 2.26). Дифференцируя зависимость Тл(х), находим
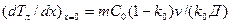 .
.
С учетом этого условия формула для определения устойчивости плоского фронта кристаллизации принимает вид
GR ³ 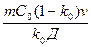 или GR/v ³
или GR/v ³ 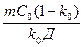 .
.
Из этих выражений видно, что любые мероприятия, направленные на увеличение коэффициента диффузии Д (перемешивание жидкости, применение ультразвука и т. п.), а также снижающие линейную скорость роста, увеличивают устойчивость плоского фронта кристаллизации.
ГЛАВА 2.7. ЯЧЕИСТАЯ И ДЕНДРИТНАЯ КРИСТАЛЛИЗАЦИЯ
На рис. 2.27 изображен фронт кристаллизации с выступами, находящимися в зоне концентрационного переохлаждения. При росте кристалла скорость перемещения фронта кристаллизации направлена по нормали к его поверхности. Скорость точки a vx будет больше, чем вертикальная компонента скорости точки b, так как точка а находится в зоне большего концентрационного переохлаждения. Поэтому в процессе роста выступ будет заостряться и увеличиваться. В точке а примесь выталкивается при кристаллизации в расплав в направлении кристаллизации. В точке b в соответствии с вектором скорости vy часть примеси будет
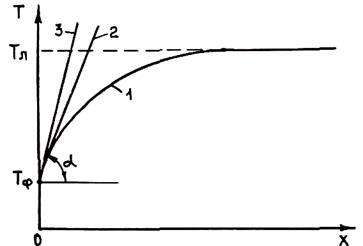
Рис. 2.26. Схема распределения температур Т(х) и Т1 (х) при отсутствии ΔTк:
1 - Тл(х); 2 – T1(х); 3 – T2(х)
выталкиваться в боковом направлении, что приводит к перемещению примеси от вершины к основанию выступов. Примесь скапливается на границах выступов (точка с на рис. 2.27). Это ведет к падению температуры ликвидуса расплава на границах выступов ниже температуры, которую имеет в данный момент расплав. В результате при продвижении фронта кристаллизации внутри периметра выступов формируются ячейки, которые не могут срастись друг с другом, пока расплав не охладится до температуры ликвидуса в этой области. Поэтому границы ячеек могут принять вид очень глубоких и узких выемок. Кристаллит будет состоять из ряда тупых выступов, связанных друг с другом на некотором расстоянии позади фронта кристаллизации.
Условие перехода от плоского фронта к ячеистой кристаллизации было получено в гл. 2.6. На рис. 2.28 приведены области ячеистой и сплошной кристаллизации, граница которых удовлетворяет этому условию. По мере уменьшения температурного градиента GR в сплавах характер фронта кристаллизации постепенно изменяется от плоского при GR ³ GRкр = mC0(1 – k0)v/(k0Д) до гексагонального ячеистого при GR < GRкр.
При дальнейшем увеличении концентрационного переохлаждения будет происходить уменьшение размеров ячеек. Так как размер ячеек не может уменьшаться безгранично, то при
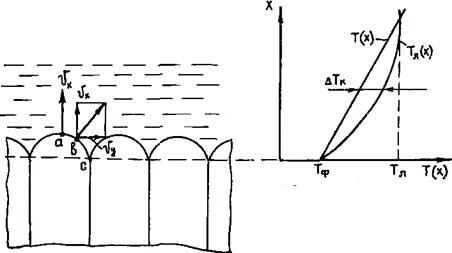
Рис. 2.27. Схема образования ячеистых кристаллов
некотором значении GR ячейки превращаются в разветвленные дендритные кристаллы. Впервые такую форму кристаллов наблюдал Д. К. Чернов, который и ввел термин «дендрит», что означает