2017-11-01
2017-11-01 2282
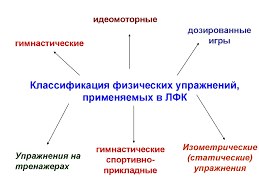
2282В предыдущих частях курса у нас показано принципиальное отличие процессов полимеризации и поликонденсации по механизму реакций. Термодинамику также целесообразно рассмотреть раздельно.
А. Полимеризация
Продолжительность процесса, степень конверсии мономера определяют производительность этого процесса. Иными словами, скорость реакций, выход продукта определяют его экономику. Всё это макроявления. Рассчитать эти макровеличины можно на основе термодинамических и кинетических законов. Термодинамика имеет дело с теплотами реакций и вообще с изменениями энергии в ходе реакции. Термодинамика определяет возможность, а кинетика-реальность осуществления процесса.
Какое же значение имеет знание термодинамики при синтезе полимеров?
Во-первых, данные о термодинамике отдельных стадий синтеза полимеров представляют собой научную основу для теплового расчёта аппаратуры и разработке энергосберегающих технологий.
В промышленности тепловой эффект полимеризации обычно выражают в кДж на 1 кг заполимеризованного мономера с учётом изменения агрегатного состояния в ходе процесса. Ориентировочные значения этих эффектов (кДж/кг) таковы: этилен-3900, пропилен -2480, формальдегид -2350, винилхлорид -1640, бутадиен -1300, изопрен - 966, стирол -670 -714. Эти значения столь существенны, что многотоннажные производства ПЭВД одновременно являются источником водяного пара высоких параметров.
Большинство процессов синтеза полимеров экзотермичны, т.е тепло выделяется, и необходимо его отводить из зоны реакции, используя те или иные технические приёмы. Часто выделяющееся тепло используется для нагрева исходной смеси.
Во-вторых, термодинамика изучает не только тепловые эффекты реакций, но и условия, при которых возможно протекание реакции в данном направлении. Возможность протекания химического процесса при данной температуре и давлении обусловлена значениями величин изменения энтальпии (Δ Н) и энтропии (Δ S) процесса, определяющих изменение термодинамического потенциала (Δ G). Для подавляющего большинства процессов полимеризации Δ Н < 0 и Δ S < 0. Из термодинамического условия осуществления реакции Δ G < 0 появляется критическая или предельная температура, выше которой полимер не может быть получен:
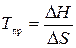
Или для растворов: Тпр.= Δ Н ∕ Δ S + R ln [ M ], где [ M ] - концентрация мономера.
Выше предельной температуры равновесие в системе мономер – полимер сдвигается в сторону мономера М ↔ П, а полимер деполимеризуется. Иными словами, ниже Тпр. образование ВМС термодинамически возможно, выше Тпр. термодинамически более выгодна деполимеризация. Это важно помнить при проектировании стадии отмывки, отпарки, нейтрализации и т.д. Например, для полиформальдегида Тпр. = 1000С, поэтому изделия из него (волокна, нетканые материалы) не подвергают высокотемпературной стирке.
Варьируя условия синтеза, т.е. переводя реакционную систему в состояние ниже Тпр. и выше Тпр., можно тонко регулировать состав получаемых продуктов, например сополимеров.
В-третьих, известно, что повышение температуры реакции обычно приводит к её ускорению. Поэтому на практике, стремясь интенсифицировать процесс синтеза, повышают его температуру. При этом ограничения налагаются обычно:
- характером зависимости молекулярной массы от температуры;
- возможностью протекания деструктивных процессов;
- тепловым взрывом, т.е. нерегулируемым повышением температуры в реакторе, имеющим место из-за превышения скорости роста производства тепла вследствие химической реакции над приростом скорости его отвода (см. часть 1 курса).
При расчёте максимальных скоростей, т.е. производительности синтеза как функции температуры реакции нужно помнить, что для обратимых процессов эта зависимость носит экстремальный характер: (рис)

W- скорость полимеризации,
Т – температура, W= 0 при Т = Тпр.
Тмак ~ 
 ,
,
Ео, Еп – энергии активации соответственно обратной и прямой реакций.
Из рисунка следует, что существует температура, при которой производительность будет максимальной, причём она связана с Δ Н
В- четвёртых, знание предельной температуры, вытекающей из основ термодинамики, чрезвычайно важно для управления процессами синтеза полимеров.
Любопытно, что возможность ограничений, связанных с приближением к предельной температуре полимеризации, стала ясна учёным и технологам только в конце 60-х годов 20 –го века при исследовании ионной полимеризации гетероатомных соединений. Именно для этих соединений значения термодинамических констант оказались таковы, что равновесии при полимеризации наступало уже при комнатной температуре.
Почему же эти явления не привлекали внимания раньше? Дело в том, что для виниловых и диеновых мономеров, которые составляли основу промышленности синтетических полимеров вместе с поликонденсационными полимерами, предельная температура полимеризации при нормальных условиях равна 250-3500С. За исключением технологических процессов полимеризации стирола в массе и этилена при высоком давлении такие высокие температуры обычно не использовались в синтезе полимеров, т.к. при них уже начинается деструкция продуктов синтеза. Известно, что величина Тпр. сильно зависит от строения молекул мономера и полимера, их агрегатного состояния. На величину предельной температуры могут значительно влиять и фазовые превращения.
Значение термодинамического потенциала μ кристаллического мономера меньше, чем аморфного. Поэтому с термодинамической точки зрения выгоднее полимеризовать аморфный мономер:
μ крист. < μаморф.
При выборе растворителя для мономера предпочтение отдаётся «худшему» растворителю (μ в этом случае больше). Изменение μ системы при полимеризации комплексно-связанных мономеров можно записать следующим образом:
Δμ = Δμ0 + Δμк.п. – Δμк.м.,где:
Δμ0- изменение химического потенциала при полимеризации в инертной среде;
Δμк.п., Δμк.м.- изменение химического потенциала соответственно полимера и мономера при комплексообразовании.
Для того, чтобы равновесие сдвигалось в сторону образования полимера, Δμк.м. должно быть больше Δμк.п,т.е. комплекс с полимером должен быть прочнее, чем комплекс с мономером.
Возможно ли изменение предельной температуры полимеризации?
Повлиять на значение предельной температуры можно, изменяя давление в реакционной системе согласно уравнению Клаузиуса-Клапейрона:

Δ V –изменение объёма системы. Известно, что Δ V/ Δ Н= 0,23~0,45 мл/кДж
Однако при полимеризации в жидкой среде эта зависимость слабая: Тпр. увеличивается на 130С на каждые 100 МПа увеличения давления, поэтому практического значения она не имеет.
Значительно важнее для практики зависимость Тпр. от концентрации мономера:
Тпр.= Δ Н ∕ Δ S + R ln [ M ], или обратная зависимость:
1/ Тпр =  ,
,
Здесь Δ Н и Δ S – стандартные величины, приведённые к одномолярному раствору мономера в данном растворителе при давлении 0,1 Мн/ м2.
В-пятых, термодинамика связана с качеством получаемого полимера. Во многих практически важных полимерных системах остаточный (непрореагировавший) мономер самым отрицательным образом влияет на свойства продукта. Его наличие может быть связано с двумя моментами:
-полимеризация имеет равновесный характер
-большая вязкость системы.
Первый момент всецело определяется термодинамикой. Второй момент определяется кинетикой. Важным показателем качества полимера является равновесное содержание мономера [ M ]∞ и степень превращения:

В условиях установившегося равновесия можно выразить общее количество молекул в смеси, а затем перейти к такому важному показателю качества полимера как ММР.
Среднечисловая степень полимеризации Рn определяется как
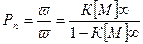 , где
, где
ω0- количество макромолекул различной длины,
ωn- количество израсходованного мономера.
Поскольку [М]∞ определяется термодинамикой системы, значит и Х и Рn тоже связаны с термодинамикой. В условиях равновесия ММР полимера будет стремиться к экспоненциальному распределению при Мw/Мn =2 (Мw-среднемассовая молекулярная масса, Мn –cреднечисловая молекулярная масса.) Это позволяет ввести в процесс приём регулирования качества полимера. Известно, что Мw/Мn служит характеристикой ширины ММР. Для теоретического анализа ММР и его связи со свойствами полимеров удобно пользоваться аналитическими, а не графическими функциями ММР. Для описания ММР используют дискретные или непрерывные функции от молекулярной массы.
Функция, характеризующая отношение числа молекул данной молекулярной массы к общему числу макромолекул, называется дискретной дифференциальной числовой функцией Рn(м) .Функция, характеризующая отношение массы молекул данной молекулярной массы ко всей массе полимера, называется дискретной дифференциальной массовой функцией ММР Рw(м)
При разных значениях Мw/Мn характер зависимостей Рп=f(j) будет различным (рис). Здесь Рп- вероятность образования макромолекул со степенью полимеризации j, j- степень полимеризации.
Известны распределения Флори, Шульца, Гаусса, Пуассона, Веслау, Бизли и др. Остановимся на зависимости 1-экспоненциальной кривой, характерной для полимеров с Мw/Мn =2
Чем же интересны эти полимеры?
При переработке и эксплуатации полимеров происходит их деструкция, приводящая в частности к изменению ММР и, следовательно, свойств продуктов. В зависимости от ширины начального ММР средние молекулярные массы могут при этом уменьшаться, если Мw/Мn <2 или увеличиваться, если Мw/Мn >2. Когда Мw/Мn =2 ММР не изменяется. Это означает, что при переработке и эксплуатации полимер сохранит совокупность свойств, заложенных при синтезе.
Б. Поликонденсация
При термодинамическом исследовании поликонденсации на первый план выступает определение зависимости молекулярной массы и др.характеристик полимера от условий проведения процесса, прежде всего таких, как температура, фазовое состояние, содержание в реакционной системе НМС и др.
В случае обратимых реакций поликонденсации после достижения системой в конце процесса равновесия состав продуктов поликонденсации полностью определяется термодинамикой протекающих реакций. Это, естественно, относится и к такому важному показателю, как равновесное содержание мономеров в продуктах реакции. В то же время состав неравновесной поликонденсационной системы зависит главным образом от кинетики процесса.
Образование полимера само по себе должно вести к уменьшению энтропии системы, т.к. мономеры как отдельные молекулы теряют способность к поступательному движению, организуясь в более упорядоченную систему. Такое изменение энтропии и наблюдается, как правило, при полимеризации. В процессе поликонденсации наряду с образованием полимера происходит выделение НМС, что ведёт к росту энтропии, причём тем в большей степени, чем выше давление пара НМС при данной температуре. Чаще всего этот эффект и определяет знак Δ S, т.е.энтропия в результате поликонденсации растёт, что относится к большей части обратимых и необратимых процессов. Естественно, что необратимая поликонденсация сопровождается несколько большим возрастанием энтропии, чем обратимая.
Очень наглядно различия проявляются в энтальпии. Обратимая поликонденсация может быть и эндо- и экзотермической, причём тепловой эффект, как правило, невелик.(не более130 кДж/моль). В то же время необратимая поликонденсация часто протекает со значительными выделениями тепла (до 340 кДж/моль). Полимеризация же обычно сопровождается уменьшением энтропии и энтальпии системы.
Для процессов поликонденсации возможны сочетания различных знаков ΔН и Δ S, поэтому общего понятия «предельная температура поликонденсации» не существует. Чаще всего Δ S поликонденсации больше нуля. Тогда при эндотермическом процессе изобарный потенциал поликонденсации будет положительным при температуре  . В этом случае можно говорить о нижней предельной температуре. Если же поликонденсация является экзотермической, и Δ S>0, то ΔG отрицательно при любой температуре.
. В этом случае можно говорить о нижней предельной температуре. Если же поликонденсация является экзотермической, и Δ S>0, то ΔG отрицательно при любой температуре.
В процессе поликонденсации кроме основных реакций: образования межзвенных связей и их разрыва при взаимодействии с НМС значительную роль могут играть реакции циклизации и межцепного обмена.
Образование циклических продуктов происходит в основном за счёт взаимодействия функциональных групп, находящихся на разных концах одной молекулы, обладающей достаточной гибкостью.
Термодинамические характеристики реакций циклизации во многомзависят от размера образующегося цикла. Макроциклы -16-тичленные и выше представляют собой ненапряжённые структуры. Поэтому тепловой эффект их образования в результате взаимодействия функциональных групп одной молекулы должен быть близок к теплоте образования межзвенной связи при реакции функциональных групп разных молекул.
Здесь Q1  Q2
Q2
Образование цикла происходит как результат столкновения двух концов молекулы олигомера или полимера. С ростом размера образующегося цикла вероятность такого столкновения уменьшается, и изменение энтропии становится всё менее благоприятным для циклообразования. Другими словами, образование больших циклов соответствует возникновению маловероятных структур, поэтому оно сопровождается снижением энтропии. Возникновение цикла значительно ограничивает свободу вращения атомов или групп атомов в молекуле, вследствие чего количество возможных конформаций становится меньше, и, соответственно уменьшается энтропия.
Отсюда следует, что вклад собственно циклообразования в термодинамические характеристики реакции в случае малых циклов сильнее влияет на ΔН, а в случае циклов больших размеров - на ΔS. Можно сделать ещё один вывод: образование в процессе поликонденсации циклических молекул термодинамически менее выгодно, чем нециклических. Энтальпийная и энтропийная составляющие ΔG более благоприятны для реакции образования нециклического продукта. Именно термодинамические факторы во многих случаях обеспечивают преимущественное образование в процессах поликонденсации полимеров линейного, а не циклического строения.
При проведении процесса поликонденсации следует учесть изменение термодинамических функций вследствие не только химических реакций, но и физических превращений (плавление, испарение, растворение, переход из аморфного состояния в кристаллическое и др.) Вклад этих процессов в термодинамические параметры поликонденсации может быть значителен.
Если проследить влияние температуры на термодинамические функции поликонденсации, то можно заметить, что в интервале температур, в которых сохраняются одни и те же агрегатные состояния реагентов, значения ΔН, ΔS и ΔG монотонно изменяются с изменением температуры, при этом чаще всего соблюдаются линейные зависимости. Очевидно, что при температурах фазовых переходов будут происходить скачки термодинамических функций, равные по величине термодинамическим функциям соответствующих переходов.
Следует отметить, что поликонденсации с образованием кристаллического полимера отвечают меньшие значения ΔН и ΔS по сравнению с процессом образования полимера в аморфном состоянии. Это связано с тем, что переход кристаллического полимера в аморфный сопровождается поглощением тепла и увеличением энтропии. Константа равновесия поликонденсации оказывается больше в случае образования кристаллического полимера, т.е. на изобарном потенциале различие в ΔН сказывается сильнее.
На термодинамические характеристики поликонденсации в растворе влияет природа растворителя. Этот эффект определяется взаимодействием растворителя со всеми участниками процесса – с мономерами, полимером и НМС. Естественно, что сильное взаимодействие полимера с растворителем может приводить к смещению равновесия в сторону образования полимера.
Использование термодинамических расчётов
Особым случаем использование термодинамических расчётов можно считать проектирование энергосберегающих процессов.
При составлении материальных и тепловых балансов при проектировании технологических установок для расчёта нагрузки оборудования инженер использует первое начало термодинамики, согласно которого внутренняя энергия изолированной системы постоянна:  , где
, где  -количество теплоты, поглощённое системой, w- работа, совершаемая системой.
-количество теплоты, поглощённое системой, w- работа, совершаемая системой.
Второе начало термодинамики, согласно которого все самопроизвольные процессы в изолированной системе происходят с возрастанием энтропии, позволяет оценить «потенциал» энергии:
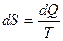 , где dQ- количество теплоты, Т- температура, т.е dSиз.>0.
, где dQ- количество теплоты, Т- температура, т.е dSиз.>0.
Разность между реальными показателями производства и потенциальными очерчивает границы возможностей по улучшению так называемого «энергетического коэффициента полезного действия». Создать идеальный с точки зрения энергетики процесс часто невозможно, но к нему можно приблизиться.
При проектировании технологических схем условия теплообмена определяются обычно компромиссом между капитальными и энергетическими затратами как функциями к.п.д. Зависимость затрат как функции к.п.д. приведена на рис..
3- общие затраты
Обычно энергетический к.п.д. в химической технологии составляет около 70%. Технологический процесс можно уподобить «чёрному ящику», в который поступают и из которого вытекают потоки энергии на разных уровнях. Энергия может, например, потребляться в виде пара, электроэнергии, а уходить в виде оборотной воды, хладоагента.
Повышение энергетической нагрузки процесса приводит к увеличению общего потребления энергии и к увеличению капиталовложений. При больших тепловых нагрузках, безусловно, нужно и более мощное теплопередающее оборудование.