 2015-05-30
2015-05-30 4099
4099Зависимость атомных радиусов от заряда ядра атома Z имеет периодический характер. В пределах одного периода с увеличением Z проявляется тенденция к уменьшению размеров атома, что особенно четко наблюдается в коротких периодах
С началом застройки нового электронного слоя, более удаленного от ядра, т. е. при переходе к следующему периоду, атомные радиусы возрастают (сравните, например, радиусы атомов фтора и натрия). В результате в пределах подгруппы с возрастанием заряда ядра размеры атомов увеличиваются.
Потеря атомов электронов приводит к уменьшению его эффективных размеров, а присоединение избыточных электронов — к увеличению. Поэтому радиус положительно заряженного иона (катиона) всегда меньше, а радиус отрицательно заряженного нона (аниона) всегда больше радиуса соответствующего электронейтрального атома.
В пределах одной подгруппы радиусы ионов одинакового заряда возрастают с увеличением заряда ядра Такая закономерность объясняется увеличением числа электронных слоев и растущим удалением внешних электронов от ядра.
Наиболее характерным химическим свойством металлов является способность их атомов легко отдавать внешние электроны и превращаться в положительно заряженные ионы, а неметаллы, наоборот, характеризуются способностью присоединять электроны с образованием отрицательных ионов. Для отрыва электрона от атома с превращением последнего в положительный ион нужно затратить некоторую энергию, называемую энергией ионизации.
Энергию ионизации можно определить путем бомбардировки атомов электронами, ускоренными в электрическом поле. То наименьшее напряжение поля, при котором скорость электронов становится достаточной для ионизации атомов, называется потенциалом ионизации атомов данного элемента и выражается в вольтах.
При затрате достаточной энергии можно оторвать от атома два, три и более электронов. Поэтому говорят о первом потенциале ионизации (энергия отрыва от атома первого электрона).втором потенциале ионизации (энергия отрыва второго электрона)
Как отмечалось выше, атомы могут не только отдавать, но и присоединять электроны. Энергия, выделяющаяся при присоединении электрона к свободному атому, называется сродством атома к электрону. Сродство к электрону, как и энергия ионизации, обычно выражается в электронвольтах. Так, сродство к электрону атома водорода равно 0,75 эВ, кислорода—1,47 эВ, фтора —3,52 эВ.
Сродство к электрону атомов металлов, как правило, близко к нулю или отрицательно; из этого следует, что для атомов большинства металлов присоединение электронов энергетически невыгодно. Сродство же к электрону атомов неметаллов всегда положительно и тем больше, чем ближе к благородному газу расположен неметалл в периодической системе; это свидетельствует об усилении неметаллических свойств по мере приближения к концу периода.
8. Химическая связь. Основные типы и характеристики химической связи. Условия и механизм ее образования. Метод валентных связей. Валентность. Понятие о методе молекулярных орбиталей.
При взаимодействии атомов между ними может возникать химическая связь, приводящая к образованию устойчивой многоатомной системы — молекулы, молекулярного нона, кристалла. условием образования химической связи является, уменьшение потенциальной энергии системы взаимодействующих атомов.
Теория химического строения. Основу теории, разработанной А. М. Бутлеровым, составляют следующие положения:
1. Атомы в молекулах соединены друг с другом в определенной последовательности. Изменение этой последовательности приводит к образованию нового вещества с новыми свойствами.
2. Соединение атомов происходит в соответствии с их валентностью.
3. Свойства веществ зависят не только от их состава, но и от их «химического строения», т. е. от порядка соединения атомов в молекулах и характера их взаимного влияния. Наиболее сильновлияют друг на друга атомы, непосредственно связанные между собой.
Представления о механизме образования химической связи, развитые Гейтлером и Лондоном на примере молекулы водорода, были распространены и на более сложные молекулы. Разработанная на этой основе теория химической связи получила название метода валентных связей (метод ВС). Метод ВС дал теоретическое объяснение важнейших свойств ковалентной связи, позволил понять строение большого числа молекул. Хотя, как мы увидим ниже, этот метод не оказался универсальным и в ряде случаев не в состоянии правильно описать структуру и свойства молекул, все же он сыграл большую роль в разработке квантово-механической теории химической связи и не потерял своего значения до настоящего времени.
Валентность — сложное понятие. Поэтому существует несколько определений валентности, выражающих различные стороны этого понятия. Наиболее общим можно считать следующее определение: валентность элемента — это способность его атомов соединяться с другими атомами в определённых соотношениях.
Первоначально за единицу валентности была принята валентность атома водорода. Валентность другого элемента можно при этом выразить числом атомов водорода, которое присоединяет к себе или замещает один атом этого другого элемента.
Мы уже знаем, что состояние электродов в атоме описывается квантовой механикой как совокупность атомных электронных орбиталей (атомных электронных облаков); каждая такая орбиталь характеризуется определенным набором атомных квантовых чисел. Метод МО исходит из предположения, что состояние электронов в молекуле также может быть описано как совокупность молекулярных электронных орбиталей (молекулярных электронных облаков), причем каждой молекулярной орбитали (МО) соответствует определенный набор молекулярных квантовых чисел. Как и в любой другой многоэлектронной системе, в молекуле сохраняет свою справедливость принцип Паули, так что на каждой МО может находиться не более двух электронов, которые должны обладать противоположно направленными спинами.
9. Водородная связь. Особые свойства воды и некоторых других соединений, способных образовывать водородную связь.
Возникновение водородной связи можно в первом приближении объяснить действием электростатических сил. Так, при образовании полярной ковалентной связи между атомом водорода и атомом фтора, который характеризуется высокой электроотрицательностью, электронное облако, первоначально принадлежавшее атому водорода, сильно смещается к атому фтора. В результате атом фтора приобретает значительный эффективный отрицательный заряд, а ядро атома водорода (протон) с «внешней» по отношению к атому фтора стороны почти лишается электронного облака. Между протоном атома водорода и отрицательно заряженным атомом фтора соседней молекулы HF возникает электростатическое притяжение, что и приводит к образованию водородной связи. Это обусловлено тем, что, обладая ничтожно малыми размерами и, в отличие от других катионов, не имея внутренних электронных слоев, которые отталкиваются отрицательно заряженными атомами, ион водорода (протон) способен проникать в электронные оболочки других атомов.
Из сказанного ясно, что условием образования водородной связи является высокая электроотрнцательность атома, непосредственно связанного в молекуле с атомом водорода. Только при этом условии электронное облако атома водорода достаточно сильно смещается в сторону атома-партнера, а последний приобретает высокий эффективный отрицательный заряд. Именно поэтому водородная связь характерна для соединения самых электроотрицательных элементов: сильнее всего она проявляется у соединений фтора и кислорода, слабее — у соединений азота и еще слабее — у соединений хлора и серы.
Энергия водородной связи значительно меньше энергии обычной ковалентной связи (150—400 кДж/моль). Она равна примерно 8 кДж/моль у соединений азота и достигает около 40 кДж/моль у соединений фтора. Однако этой энергии достаточно, чтобы вызвать ассоциацию молекул, т. е. их объединение в димеры (удвоенные молекулы) или полимеры, которые в ряде случаев существуют не только в жидком состоянии вещества, но сохраняются и при переходе его в пар. Именно ассоциация молекул, затрудняющая отрыв их друг от друга, и служит причиной аномально высоких температур плавления и кипения таких веществ, как фтороводород, вода, аммиак. Другие особенности этих веществ, обусловленные образованием водородных связей и ассоциацией молекул, будут рассмотрены ниже, при изучении отдельных соединений.
Водородная связь служит причиной некоторых важных особенностей воды — вещества, играющего огромную роль в процессах, протекающих в живой и неживой природе. Она в значительной мере определяет свойства и таких биологически важных веществ, как белки и нуклеиновые кислоты.
10.Донорно-акдепторная связь. Комплексные соединения. Комплексообразователь и лиганды. Заряд комплексообразователя и координационной сферы. Координационное число константы нестойкости.
Знакомясь с элементами подгруппы меди, мы видели, что ионы этих элементов способны присоединять к себе другие ионы или нейтральные молекулы (например, NH3), образуя более сложные комплексные ионы. При связывании последних ионами противоположного знака получаются различные комплексные соединения.
Комплексные соединения составляют наиболее обширный и разнообразный класс неорганических веществ. К ним принадлежат также многие металлорганические соединения (см. § 163), связывающие воедино ранее разобщенные неорганическую химию и органическую химию. Многие комплексные соединения —витамин Bi2, гемоглобин, хлорофилл и другие — играют большую роль в физиологических и биохимических процессах. Согласно координационной теории, в молекуле любого комплексного соединения один из ионов, обычно положительно заряженный, занимает центральное место и называется комплексообразователем или центральным ионом. Вокруг него в непосредственной близости расположено или, как говорят, координировано некоторое число противоположно заряженных ионов или электронейтральных молекул, называемых лигандами (или аддендами) и образующих внутреннюю координационную сферу соединения. Остальные ионы, не разместившиеся во внутренней сфере, находятся на более далеком расстоянии от центрального иона, составляя внешнюю координационную сферу. Число лигандов, окружающих центральный ион, называется «координационным числом».
Заряд комплексного иона равен алгебраической сумме зарядов составляющих его простых ионов
[Ag(NH3)2]+ + I ------> AgI + 2NH,
2[Ag(NH3)2]+ + H2S —> Ag2S +2NH,+2NH4+
Диссоциация ионов [Ag(NH3)2]+, согласно приведенному выше уравнению, как и диссоциация всякого слабого электролита, подчиняется закону действия масс и может быть охарактеризована соответствующей константой равновесия, называемой константой нестойкости комплексного иона:
11.Межмолекулярное взаимодействие. Ван-дер-Ваальсовы силы. Типы межмолекулярного взаимодействия (ориентационное, индукционное идисперсионное). Влияние сил межмолекулярного взаимодействия на свойства веществ.
Силы, удерживающие частицы жидкости или твердого тела друг около друга,
имеют электрическую природу. Но в зависимости от того, что представляют собой частицы —являются ли они атомами металлического или неметаллического элемента, ионами или молекулами, — эти силы существенно различны.
Если вещество построено из атомов, но не является металлом, то его атомы обычно связаны друг с другом ковалентной связью. Если вещество — металл, то часть электронов его атомов становятся общими для всех атомов; эти электроны свободно движутся между атомами, связывая их друг с другом. Если вещество имеет ионное строение, то образующие его ионы удерживаются друг около друга силами электростатического притяжения. О ковалентной и ионной связи говорилось в главе IV. О связи между частицами в металлах рассказывается в главе XVI. В веществах с молекулярной структурой имеет место межмолекулярное взаимодействие.
Силы межмолекулярного взаимодействия, называемые также силами Ван-дер-Ваальса, слабее ковалентных сил, но проявляются на больших расстояниях. В основе их лежит электростатическое взаимодействие диполей, но в различных веществах механизм возникновения диполей различен. Если вещество состоит из полярных молекул, например, молекул Н 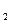 О или НС1, то в конденсированном состоянии соседние молекулярные диполи ориентируются друг по отношению к другу противоположно заряженными полюсами, вследствие чего наблюдается их взаимное притяжение. Такой вид межмолекулярного взаимодействия называется ориентационным взаимодействием. Тепловое движение молекул препятствует взаимной ориентации молекул, поэтому с ростом температуры ориентационный эффект ослабевает.
О или НС1, то в конденсированном состоянии соседние молекулярные диполи ориентируются друг по отношению к другу противоположно заряженными полюсами, вследствие чего наблюдается их взаимное притяжение. Такой вид межмолекулярного взаимодействия называется ориентационным взаимодействием. Тепловое движение молекул препятствует взаимной ориентации молекул, поэтому с ростом температуры ориентационный эффект ослабевает.
В случае веществ, состоящих из неполярных, но способных к поляризации молекул, например, СО , наблюдается возникновение наведенных или индуцированных диполей. Причина их появления обычно состоит в том, что каждый атом создает вблизи себя электрическое поле, оказывающее поляризующее действие на ближайший атом соседней молекулы. Молекула поляризуется, и образовавшийся индуцированный диполь в свою очередь поляризует соседние молекулы. В результате происходит взаимное притяжение молекул друг к другу. Это индукционное взаимодействие наблюдается также и у веществ с полярными молекулами, но при этом оно обычно значительно слабее ориентационного.
Относительная величина рассмотренных видов межмолекулярных сил зависит от полярности и от поляризуемости молекул вещества. Чем больше полярность молекул, тем больше ориентационные силы. Чем больше деформируемость, тем слабее связаны внешние электроны атомов, т. е. чем эти атомы крупнее, тем значительнее дисперсионные силы. Таким образом, в ряду однотипных веществ дисперсионное взаимодействие возрастает с увеличением размеров атомов, составляющих молекулы этих веществ. Например, в случае НС1 на долю дисперсионных сил приходится 81 % всего межмолекулярного взаимодействия, для НВr эта величина составляет 95%, а для HI 99,5%. Индукционные силы почти всегда малы.
12.Химическая термодинамика. Энергетика химических процессов. Внутренняя энергия и энтальпия. Термохимические уравнения. Теплота образования и разложения веществ.
Если при образовании какого-либо соединения выделяется {или поглощается) некоторое количество теплоты, то при разложении этого соединения в тех же условиях такое же количество теплоты поглощается (или выделяется).
Количество теплоты, которое выделяется при образовании одного моля соединения из простых веществ, называется теплотой образования данного соединения.
Химические уравнения, в которых указано количество выделяющейся или поглощаемой теплоты, называются термохимическими уравнениями. Величина теплового эффекта указывается обычно в правой части уравнения со знаком плюс в случае экзотермической реакции и со знаком минус в случае эндотермической реакции.
Внутренняя энергия U вещества (или системы) — это полная энергия частиц, составляющих данное вещество (см. также § 54). Она слагается из кинетической и потенциальной энергий частиц. Кинетическая энергия — это энергия поступательного, колебательного и вращательного движения частиц; потенциальная энергия обусловлена силами притяжения и отталкивания, действующими между частицами.
Энтальпия. Однако чаще в химии приходится иметь дело с процессами, протекающими при постоянном давлении. При этом удобно пользоваться величиной энтальпии Н, определяемой соотношением:
Н= U + PV
изменение энтальпии равно взятому с обратным знаком тепловому эффекту реакции, проведенной при постоянной температуре и постоянном давлении
13.Стандартные тепловые эффекты различных процессов. Основной закон термохимии (закон Гесса). Применение термохимических расчетов,
Закона Гесса: суммарный тепловой эффект некоторого ряда последовательных реакций равен суммарному тепловому эффекту любого другого ряда реакций, если одинаковы исходные в-ва, их кол-ва и состояние, а также одинаковы продукты реакций и их кол-во и состояние.
Следствие 1: тепловой эффект разложения хим. соединения до простых в-в равен тепловому эффекту образования этого соединения из этих простых в-в, взятому с обратным знаком.
Следствие 2: тепловой эффект реакции равен разности между суммой теплот образования продуктов реакции и суммой теплот образования исх. в-в.
Разность ТЭ двух реакций, начальное состояние в-в у которого различны, а конечные одинаковы, представляют собой ТЭ перехода в-в из одного начальное состояние в другое.
Разность ТЭ одной и той же р-ции, для которой состояние в-в одинаковы, а конечных – различны представляют собой ТЭ перехода из одного конечного состояния в другое.
14.Химическое сродство. Энтропия. Ее изменение при химических процессах. Стандартные энтропии веществ. Методы расчета" изменения энтропии в ходе химической реакции.
Энтропия-мера беспорядка.

 Энтропия (S) связана с числом (W) равновероятных микроскопических состояний, которыми можно реализовать данное макроскопическое состояние системы, уравнением s=h/t Наименьшую энтропию имеют идеально правильно построенные кристаллы при абсолютном нуле. Энтропия кристалла, в структуре которого имеются какие-либо неправильности, уже при абсолютном нуле несколько больше, так как нарушения идеальности могут реализоваться не единственным способом. С повышением температуры энтропия всегда возрастает, так как возрастает интенсивность движения частиц, а следовательно, растет число способов их расположения. Возрастает она также при превращении вещества из кристаллического состояния в жидкое и, в особенности, при переходе из жидкого состояния в газообразное. Если процесс проводится обратимо и при постоянной температуре (изотермически), то изменение энтропии связано с поглощаемой теплотой уравнением
Энтропия (S) связана с числом (W) равновероятных микроскопических состояний, которыми можно реализовать данное макроскопическое состояние системы, уравнением s=h/t Наименьшую энтропию имеют идеально правильно построенные кристаллы при абсолютном нуле. Энтропия кристалла, в структуре которого имеются какие-либо неправильности, уже при абсолютном нуле несколько больше, так как нарушения идеальности могут реализоваться не единственным способом. С повышением температуры энтропия всегда возрастает, так как возрастает интенсивность движения частиц, а следовательно, растет число способов их расположения. Возрастает она также при превращении вещества из кристаллического состояния в жидкое и, в особенности, при переходе из жидкого состояния в газообразное. Если процесс проводится обратимо и при постоянной температуре (изотермически), то изменение энтропии связано с поглощаемой теплотой уравнением
S =Qo6P/T
где Q06p — количество теплоты, поглощенной системой в изотермическом обратимом процессе; Т — абсолютная температура.
С помощью этого уравнения можно определить, например, изменение энтропии при плавлении и кипении веществ.
По величине стандартной энтропии можно делать некоторые выводы о свойствах вещ-в: чем больше атомов сожержется в молекуле вещ-ва, тем его энтропия выше.
Для качественного определения изменения энтропии должны знать, как изменится число молей газа (твёрдые и жидкие вещ-ва не учитываем), т. К. их энтропия очень мала). Для твёрдых тел энтропия амофорных тел выше,чем энтропия тех же тел кристаллических: SiO2-амофорное-стекло, кристаллическое-кварц.
Изобрано-изотермический потенциал. Принципиальная возможность или невозможность осуществления процесса. Энтальпийный и энтропийный факторы и направление процесса Расчет направления протекания химических реакций.
Как показывается в термодинамике, можно ввести такие функции, которые отражают влияние на направление протекания процесса как тенденции к уменьшению внутренней энергии, так и тенденции к достижению наиболее вероятного состояния системы. Знак изменения подобной функции при той или иной реакции может служить критерием возможности самопроизвольного протекания реакции. Для изотермических реакций, протекающих при постоянном давлении, такой функцией является энергия Гиббса G, называемая также изобарно-изотермическим потенциалом, изобарным потенциалом или свободной энергией при постоянном давлении.
Рассмотренное нами явление расширения газа представляет собой пример проявления принципа направленности процессов к наиболее вероятному состоянию, т. е. к состоянию, которому соответствует максимальная беспорядочность распределения частиц. Направление самопроизвольного протекания химических реакций и определяется совокупным действием двух факторов: тенденцией к переходу системы в состояние с наименьшей внутренней энергией и тенденцией к достижению наиболее вероятного состояния. Согласно закону Гесса, стандартное изменение энтальпии реакции (сокращенно: стандартная энтальпия реакции) равно сумме стандартных энтальпий образования продуктов реакции за вычетом  суммы стандартных энтальпий образования исходных веществ.
суммы стандартных энтальпий образования исходных веществ.
Аналогично стандартное изменение энергии Гиббса реакции (сокращенно: стандартная энергия Гиббса реакции) равно сумме стандартных энергий Гиббса образования продуктов реакции за вычетом суммы стандартных энергий Гиббса образования исходных веществ. При этом все суммирования производятся с учетом числа молей участвующих в реакции веществ в соответствии с ее уравнением.

