2015-06-26
2015-06-26 7370
7370Ферментопатии − это заболевания, развивающиеся в результате наследственного дефицита некоторых ферментов или вследствие приобретенной их недостаточности. Известно более 400 энзимопатий, связанных с нарушением обмена углеводов, липидов, белков и аминокислот, азотистых оснований, порфиринов.
Энзимопатии могут приводить к нарушению важнейших метаболических путей, блокированию синтеза жизненно важных метаболитов и продуктов, накоплению в жидкостях и тканях организма субстратов и промежуточных продуктов реакций, которые в ряде случаев оказывают токсическое действие на организм.
Примером в р о ж д е н н ы х н а р у ш е н и й м е т а б о л и з м а у г л е в о д о в могут служить гликогенозы и агликогенозы, характеризующиеся либо отложением в тканях больших количеств гликогена, необычных его видов, либо нарушением синтеза гликогена. Причиной гликогенозов служит дефицит или полное отсутствие ферментов, катализирующих процесс распада гликогена. Агликогенозы возникают при отсутствии ферментов синтеза гликогена.
В случае дефицита или отсутствия ряда лизосомных ферментов, принимающих участие в обмене гликопротеинов, протеогликанов и гликолипидов, возникают заболевания гликозидозы, мукополисахаридозы, гликолипидозы соответственно. Они характеризуются накоплением в лизосомах почти всех тканей продуктов неполного расщепления гетерополисахаридов и гликозаминогликанов. Общими признаками разных форм таких заболеваний являются изменение скелета, деформация суставов, поражение печени, селезенки, сердца, кровеносных сосудов, задержка умственного и физического развития.
К энзимопатиям углеводного обмена относятся также такие заболевания, как галактоземия, эссенциальная фруктозурия, наследственная неперено-симость фруктозы, недостаточность дисахаридаз.
Причиной галактоземии может быть дефект одного из трех ферментов: галактокиназы, галактозо-1-фосфатуридилтрансферазы и УДФ-галактозо-4-эпимеразы. При дефекте галактокиназы повышается концентрация галактозы в крови, что сопровождается галактозурией. В результате образования катаракты ухудшается зрение.
Наиболее изученным является дефицит галактозо-1-фосфатуридил-трансферазы. Заболевание проявляется уже в первые дни и недели после рождения. Новорожденные неохотно принимают молоко, наблюдается рвота, вздутый живот, диспепсия, гипогликемия, печень и селезенка увеличиваются. Катаракта проявляется не ранее 3-й недели и постепенно приводит к полной слепоте. Из питания необходимо исключить продукты, имеющие в своем составе галактозу (в первую очередь молоко, содержащее дисахарид лактозу).
Эссенциальная (семейная) фруктозурия обусловлена дефицитом фермента фруктокиназы, катализирующего превращение фруктозы во фруктозо-1-фосфат. Поступающая в организм фруктоза не фосфорилируется, накапливается в крови и выводится с мочой.
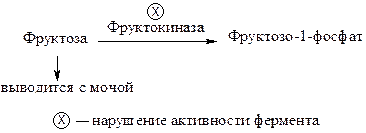
Накапливающийся при этом субстрат (фруктоза) не является токсичным для организма, продукт блокированной реакции (фруктозо-1-фосфат) может синтезироваться обходным путем. Такое нарушение не приводит к тяжелым последствиям. Семейная фруктозурия протекает бессимптомно и обнаруживается случайно.
Более тяжелым нарушением является врожденная (наследственная) непереносимость фруктозы. Причиной заболевания является дефицит фермента фруктозо-1-фосфатальдолазы, катализирующего реакцию расщепления фруктозо-1-фосфата на глицеральдегид и диоксиацетонфосфат:
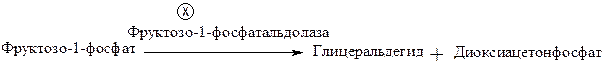
Субстрат (фруктозо-1-фосфат) накапливается в организме и оказывает токсическое действие, в частности, тормозит активность фосфоглюкомутазы, превращающей глюкозо-1-фосфат в глюкозо-6-фосфат. Это приводит к торможению распада гликогена на этапе глюкозо-6-фосфата и последующему развитию гипогликемии, усилению мобилизации липидов. При переводе детей на смешанное или искусственное питание после приема фруктового сока и фруктов возникают слабость, дрожь, судороги, потливость, рвота, нарушение сознания. Непереносимость фруктозы протекает тяжело и способна вызвать смертельный исход.
При редком наследственном заболевании эссенциальной пентозурии в моче появляется большое количество L -ксилулозы (до 4 г / сутки), обусловлено дефектом фермента L -ксилулозоредуктазы, необходимого для восстановления L -ксилулозы в ксилитол. Существенные клинические проявления отсутствуют.
Недостаточность дисахаридаз (лактазы, сахаразы, мальтазы и изомальтазы) приводит к нарушению кишечного всасывания дисахаридов, диспепсии, усилению процессов брожения в кишечнике, усилению перистальтики и нарушению всасывания воды. При врожденном дефиците лактазы симптомы появляются уже после первых прикладываний новорожденных к груди. Взрослые люди с недостатком лактазы испытывают метеоризм и диарею в результате употребления молока.
К энзимопатиям с в р о ж д е н н ы м и н а р у ш е н и я м и л и п и д- н о г о о б м е н а относятся сфинголипидозы, болезнь Рефсума, гиперпропионемия, метилмалоновая ацидурия и др.
Сфинголипидозы возникают из-за отсутствия лизосомных ферментов, которые принимают участие в процессе распада сфинголипидов, что влияет на процессы развития структуры центральной нервной системы и может привести к летальному исходу. При сфинголипидозах могут наблюдаться дефекты b -глюкозидазы, сфингомиелиназы, a - и b -галактозидаз и других ферментов. Например, болезнь Нимана − Пика обусловлена наследственным дефектом сфингомиелиназы. Наблюдается увеличение печени и селезенки, умственная отсталость, летальный исход в раннем возрасте.
Болезнь Рефсума вызвана нарушением b -окисления разветвленных жирных кислот, особенно фитановой кислоты, образующейся при окислении фитола. Фитановая кислота накапливается в плазме крови, где ее содержание достигает 20 % содержания всех жирных кислот. При данной патологии следует избегать употребления в пищу веществ, содержащих фитол (особенно молоко).
Гиперпропионемия возникает вследствие дефекта пропионил-КоА-карбоксилазы. В крови накапливается пропионовая кислота, которая выводится в больших количествах с мочой в виде натриевой соли. Состояние улучшается при введении больших доз биотина.
Причиной метилмалоновой ацидурии является дефект метилмалонил-КоА-мутазы. В этом случае нарушается превращение метилмалонил-КоА в сукцинил-КоА. Метилмалонат накапливается в крови и выделяется с мочой. В некоторых случаях состояние удается улучшить назначением больших доз витамина В12.
Недостаточность липопротеинлипазы проявляется в детстве симптомами, указывающими на накопление жиров в различных органах и тканях: в коже (ксантоматоз), в печени (гепатомегалия), а также болями в области живота (симптом, сопровождающий гиперхиломикронемию).
Дефект ЛХАТ приводит к накоплению неэтерифицированного холестерина в тканях, следствием чего является преждевременное развитие атеросклероза, помутнение роговой оболочки глаз, повреждение почек, анемия.
При генетическом дефекте карнитин-ацилтрансферазы нарушается процесс b- окисления жирных кислот, являющийся важнейшим источником энергии в работающих мышцах в аэробных условиях.
Скорость b- окисления жирных кислот может снизиться также вследствие дефекта фермента ацил-КоА-дегидрогеназы. Для детей грудного возраста в отличие от взрослых b -окисления жирных кислот, получаемых с грудным молоком, является основным источником энергии. У детей с дефектом данного фермента уменьшается скорость b -окисления жирных кислот, глюкоза используется в большей степени, чем при нормальном метаболизме, в результате развивается гипогликемия. Это состояние является внезапной причиной детской смертности более чем в 10 % случаев.
Э н з и м о п а т и и, п р и в о д я щ и е к н а р у ш е н и ю о б м е н а б е л к о в и а м и н о к и с л о т, относятся к довольно распространенным видам наследственных нарушений метаболизма. При наследственном заболевании фенилкетонурии (фенилпировиноградной олигофрении) нарушается превращение фенилаланина в тирозин вследствие дефекта фермента фенилаланин-4-монооксигеназы. В крови повышается концентрация фенилаланина и побочных продуктов его превращения − фенилпирувата и фениллактата, токсичных для клеток мозга. Кроме того, возникает дефицит нейромедиатора норадреналина в нервной ткани, предшественником которого является тирозин:
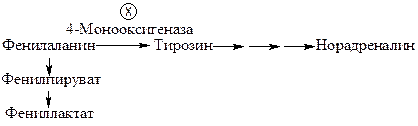
Частота фенилкетонурии новоржденных в мире составляет от 1:10 000 до 1:20 000. Наиболее частая форма классической фенилкетонурии проявляется характерным симптомокомплексом: умственная отсталость, судорожный синдром, склонность к экземе, нарушение пигментного обмена. От больных исходит специфический «мышиный» запах, обусловленный высоким содержанием фенольных кислот в биологических жидкостях. Симптомы появляются в первый год жизни. Больные при отсутствии лечения, как правило, не доживают до тридцати лет. При подтверждении диагноза фенилкетонурии новорожденным назначают диету с низким содержанием фенилаланина для предотвращения развития клинических симптомов. Практика показала, что соблюдение указанной диеты до восьмилетнего возраста дает положительные результаты, поскольку за этот период заканчивается процесс миелинизации мозга. Накопление фенилаланина в клетках мозга нарушает реакции синтеза цереброзидсульфатов, участвующих в защите мозга от демиелинизации.
Алкаптонурия − заболевание, обусловленное дефектом фермента гомогентизат-1,2-диоксигеназы, участвующего в процессе обмена тирозина. В крови накапливается гомогентизиновая кислота, которая выводится с мочой; при выдерживании на воздухе моча темнеет вследствие образования черного пигмента алкаптона.
Известно около двух десятков наследственных заболеваний, обусловленных нарушением обмена аминокислот. Диагностика таких энзимопатий возможна только с помощью методов клинической энзимологии.
Н а р у ш е н и я о б м е н а п у р и н о в ы х и п и р и м и д и н о в ы х о с н о в а н и й, обусловленные энзимопатиями, приводят к развитию таких патологий, как синдром Леша − Нихена, оротовая ацидурия, иммуно-дефицитные состояния.
Синдром Леша – Нихена – наследственное заболевание, причиной которого является дефект фермента гуанингипоксантинфосфорибозил-трансферазы, катализирующего превращение гуанина в ГМФ и гипоксантина в ИМФ. Заболевание клинически проявляется церебральным параличом, умственной отсталостью, аутоагрессией, членовредительством, выраженной гиперурикемией (повышением концентрации мочевой кислоты в плазме крови).
При генетическом дефекте аденозиндезаминазы у детей в период до двух лет наблюдается тяжелый комбинированный иммунодефицит, связанный с нарушениями созревания и пролиферации Т- и В-лимфоцитов, со слабым иммунным ответом на антиген (уменьшение образования плазматических клеток). Дефект аденозиндезаминазы выявляется во многих тканях, но патологические последствия развиваются главным образом в лимфоцитах. Недоразвиты тимус и лимфатические узлы. Дети предрасположены к бактериальным, вирусным и грибковым болезням, обычно умирают к двухлетнему возрасту в результате повреждения бронхолегочной системы и дисплазии костного мозга.
Врожденная оротовая ацидурия характеризуется снижением активности двух ферментов: оротатфосфорибозилтрансферазы или оротидилатдекарбоксилазы. Развивается дефицит пиримидиновых нуклеотидов, так как в организме нет обходных путей их синтеза. В детском возрасте для больных характерны отставание в умственном и физическом развитии, мегалобластическая анемия, накопление и выделение с мочой оротовой кислоты. Заболевание легко поддается лечению уридином. При отсутствии лечения пациенты подвергаются частым инфекциям.
Наследственные заболевания, обусловленные н е д о с т а т о ч н о с т ь ю ф е р м е н т о в, у ч а с т в у ю щ и х в м е т а б о л и з м е п о р ф и р и н о в и с и н т е з е г е м а, называются порфириями. Существует несколько типов порфирий, характеризующихся накоплением в организме и повышенным выделением порфиринов и их предшественников.
Известны н а с л е д с т в е н н ы е м е т а б о л и ч е с к и е н а р у ш е н и я, обусловленные недостатком каждого из пяти ферментов, катализирую-щих в печени отдельные стадии процесса с и н т е з а м о ч е в и н ы. Поскольку в цикле мочевинообразования аммиак превращается в нетоксичную мочевину, все нарушения ее синтеза вызывают аммиачное отравление. Клиническими симптомами является рвота (у детей), отвращение к богатым белками продуктам, нарушение координации движений, раздражительность, сонливость, умственная отсталость. Значительное улучшение наблюдается при ограничении белка в диете, при этом могут быть предотвращены нарушения мозговой деятельности.
Уровень активности ферментов в плазме крови находится под контролем естественных ингибиторов белковой природы. Обнаружен наследственный дефицит белковых ингибиторов протеолиза (в частности, a1 -анти-трипсина и a1- антипротеиназы), приводящий к развитию заболеваний бронхолегочного дерева и неонатального гепатита, который способен вызвать цирроз печени. a1 -Антитрипсин является ингибитором трипсина и других протеаз, которые с места своего образования (клетки поджелудочной железы) могут попадать в кровь. Недостаточность a1 -антитрипсина и
a1- антипротеиназы делает возможным расщепление структур соединительной ткани под действием трипсина и эластазы, причем особенно поражается соединительная ткань легких, где прежде всего разрушаются межальвеолярные перегородки. В легких вместо отдельных альвеол образуются обширные полости, способность которых к газообмену снижена по причине малой площади поверхности. Развивается эмфизема легких, в том числе и у молодых людей (20 − 30 лет). Усугубляющим фактором при этом является курение, поскольку компоненты табачного дыма окисляют тиоловую группу цистеина в активном участке молекулы a1 -антитрипсина, что снижает активность присутствующего в небольшом количестве фермента.