 2018-01-21
2018-01-21 8170
8170химическая реакция, сопровождающаяся выделением теплоты. Э. р. являются, например, Горение,Нейтрализация, большинство реакций образования химических соединений из простых веществ Количество выделяющейся при Э. р. теплоты зависит от массы реагентов и их природы, агрегатного состояния исходных веществ и продуктов взаимодействия, типа реакции и условий её осуществления (температуры, давления и др.). По тепловому эффекту Э. р. противоположны эндотермическим реакциям.
Эндотерми́ческие реа́кции (от др.-греч. ἔνδον — внутри и θέρμη — тепло) — химические реакции, сопровождающиеся поглощением теплоты. Для эндотермических реакций изменение энтальпии и внутренней энергии имеют положительные значения ( ,
,  ), таким образом, продукты реакции содержат больше энергии, чем исходные компоненты.
), таким образом, продукты реакции содержат больше энергии, чем исходные компоненты.
К эндотермическим реакциям относятся:
· реакции восстановления металлов из оксидов,
· электролиза (поглощается электрическая энергия),
· электролитической диссоциации (например, растворение солей в воде),
· ионизации,
· фотосинтеза.
Эндотермические реакции противоположны экзотермическим реакциям.
Любая химическая реакция сопровождается выделением или поглощением энергии. Особенность термохимических уравнений заключается в том, что при работе с ними можно переносить формулы веществ и величины тепловых эффектов из одной части уравнения в другую. С обычными уравнениями химических реакций так поступать, как правило, нельзя.
47) Станда́ртные состоя́ния — в химической термодинамике условно принятые состояния индивидуальных веществ и компонентов растворов при оценке термодинамических величин.
Необходимость введения «стандартных состояний» связана с тем, что термодинамические закономерности не описывают достаточно точно поведение реальных веществ, когда количественной характеристикой служит давление или концентрация. Стандартные состояния выбирают из соображений удобства расчётов, и они могут меняться при переходе от одной задачи к другой.
В стандартных состояниях значения термодинамических величин называют «стандартными» и обозначают нулем в верхнем индексе[1], например: G0, H0, m0 — это соответственно стандартные энергия Гиббса, энтальпия, химический потенциал вещества. Вместо давления в термодинамических уравнениях для идеальных газов и растворов используютфугитивность (летучесть), а вместо концентрации — активность.
Стандартная энтальпия химической связи — это изменение энтальпии в реакции образования одного моля двухатомных молекул (или других двухатомных частиц) из атомов веществ, находящихся в газообразном состоянии:
H(г) + Cl(г) = HCl(г)
С(г) + H(г) = CH(г)
Энтальпия (теплота) образования - это тепловой эффект реакции образования 1 моль сложного вещества из простых веществ: DfH [Дж/моль; кДж/моль]. Обычно в расчетах используют стандартные энтальпии образования.СТАНДАРТНАЯ ЭНТАЛЬПИЯ ОБРАЗОВАНИЯ DfH°(298 K) это тепловой эффект образования 1 моль сложного вещества из простых веществ при стандартных условиях (T = 298,15 K и p = 101,3 кПа). Стандартные энтальпии образования простых веществ, устойчивых в стандартных условиях (газообразный O2, кристаллический I2 и т.д.) принимают равными нулю. Например, окисление водорода можно представить тремя уравнениями:
(6) 2H2(г) + O2(г) = 2H2O(ж); DH°(298K)6 = -571,6 кДж
(7) H2(г) + 1/2O2(г) = H2O(ж); DH°(298K)7 = -285,8 кДж
(8) H2(г) + 1/2O2(г) = H2O(г); DH°(298K)8= -241,8 кДж
Каждому уравнению соответствует определенное значение теплового эффекта. И только тепловой эффект реакции, описываемой уравнением (7), будет равен стандартной теплоте образования воды DH°(298 K)7 = DfH°(298 K, H2O(ж)). Согласно этому уравнению в реакции образуется 1 моль воды, стандартным состоянием которой при 298 K является жидкое.
Для многих веществ стандартные теплоты образования известны и сведены в справочные таблицы.
Теплоёмкость тела (обычно обозначается латинской буквой C) — физическая величина, определяющая отношение бесконечно малого количества теплоты δ Q, полученного телом, к соответствующему приращению его температуры δ T:

Единица измерения теплоёмкости в системе СИ — Дж/К.
Удельной теплоёмкостью называется теплоёмкость, отнесённая к единичному количеству вещества. Количество вещества может быть измерено в килограммах, кубических метрах и молях. В зависимости от того, к какой количественной единице относится теплоёмкость, различают массовую, объёмную и молярную теплоёмкость.
Массовая теплоёмкость (С) — это количество теплоты, которое необходимо подвести к единице массы вещества, чтобы нагреть его на единицу температуры. В СИ измеряется в джоулях на килограмм на кельвин (Дж·кг−1·К−1).
Объёмная теплоёмкость (С′) — это количество теплоты, которое необходимо подвести к единице объёма вещества, чтобы нагреть его на единицу температуры. В СИ измеряется в джоулях на кубический метр на кельвин (Дж·м−3·К−1).
Молярная теплоёмкость (С μ) — это количество теплоты, которое необходимо подвести к 1 молю вещества, чтобы нагреть его на единицу температуры. В СИ измеряется в джоулях на моль на кельвин (Дж/(моль·К)).
Гесса закон
Гесса закон
основной закон термохимии, согласно которому тепловой эффект реакции зависит лишь от начального и конечного состояний системы и не зависит от промежуточных состояний и путей перехода. Г. з. был открыт Г. И. Гессом в 1840 на основе экспериментальных исследований. Он представляет собой одну из форм позднее открытого закона сохранения энергии в применении его к химическим реакциям и относится к процессам, происходящим при постоянном объёме или при постоянном давлении. Г. з. широко используется для определения расчётным путём теплового эффекта интересующего процесса на основе экспериментальных данных, относящихся к др. процессам (в т. ч. даже к процессам, практически недоступным в данных условиях). Так, для 298,15 К теплоту образования окиси углерода ( ккал/моль) из графита можно рассчитать, зная, что теплоты сгорания
ккал/моль) из графита можно рассчитать, зная, что теплоты сгорания 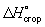 2 при этой температуре равны соответственно —282,99 и —393,32кдж/моль(—67,635 и—94,051ккал/моль). Рассматривая два пути образования CO2из графита при непосредственном сжигании его до CO2и при промежуточном образовании СО (см. рис.) и зная, что по Г. з. общий тепловой эффект обоих путей перехода должен быть одинаковым, находим
2 при этой температуре равны соответственно —282,99 и —393,32кдж/моль(—67,635 и—94,051ккал/моль). Рассматривая два пути образования CO2из графита при непосредственном сжигании его до CO2и при промежуточном образовании СО (см. рис.) и зная, что по Г. з. общий тепловой эффект обоих путей перехода должен быть одинаковым, находим  ккал/моль (теплота выделяется).
ккал/моль (теплота выделяется).
49) Понятие энтропии было введено Р. Клаузиусом[2], сформулировавшим второе начало термодинамики, согласно которому переход теплоты от более холодного тела к более теплому не может происходить без затраты внешней работы.
Он определил изменение энтропии термодинамической системы при обратимом процессе как отношение изменения общего количества тепла ΔQ к величине абсолютной температуры T:
Рудольф Клаузиус дал величине S имя «энтропия», происходящее от греческого слова τρoπή, «изменение» (изменение, превращение, преобразование).
Эта формула применима только для изотермического процесса (происходящего при постоянной температуре). Её обобщение на случай произвольного квазистатического процесса выглядит так:
,
где dS - приращение (дифференциал) энтропии, а δQ - бесконечно малое приращение количества теплоты.
Заметим, что энтропия является функцией состояния, поэтому в левой части равенства стоит её полный дифференциал. Напротив, количество теплоты является функцией процесса, в котором эта теплота была передана, поэтому δQ ни в коем случае нельзя считать полным дифференциалом.
Энтропия, таким образом, определена вплоть до произвольной аддитивной постоянной. Третье начало термодинамики позволяет определить её точно: при этом энтропию равновесной системы при абсолютном нуле температуры считают равной нулю.
Энтропия — это количественная мера той теплоты, которая не переходит в работу.
S2 - S1 = ΔS =
Или, другими словами, энтропия — мера рассеивания свободной энергии. А ведь нам уже известно, что любая открытая термодинамическая система в стационарном состоянии стремится к минимальному рассеиванию свободной энергии. Поэтому если в силу причин система отклонилась от стационарного состояния, то вследствие стремления системы к минимальной энтропии, в ней возникают внутренние изменения, возвращающие ее в стационарное состояние.
Второй закон
 | Энтропия изолированная система не в равновесии имеет тенденцию увеличиваться с течением времени, приближаясь к максимальному значению в равновесии. |  |
Третий закон
| | При стремлении температуры к абсолютному нулю, энтропия системы приближается к постоянному минимуму. | |
Короче говоря, постулируется, что энтропия — «температурный иждивенец» и приводит к формулировке идеиабсолютного нуля.
ГИББСА ЭНЕРГИЯ
ГИББСА ЭНЕРГИЯ
(изобарно-изотермический потенциал, свободная энтальпия), один из потенциалов термодинамических; характеристическая функциятермодинамич. системы при независимых параметрах р (давление), Т (термодинамич. темп-pa) и N (число ч-ц в системе). Обозначается G (иногда Z, Ф), определяется через энтальпию Н, энтропию S и темп-ру Т равенством: G=H-TS. С Гельмгольца энергией F Г. э. связана соотношением: G=F+pV. Г. э. пропорц. числу ч-ц N; отнесённая к одной ч-це, она наз. химическим потенциалом. Г. э. удобна для описания процессов, в к-рых возможен обмен в-вом с окружающими телами. Понятие «Г. э.» введено в термодинамику амер. физиком Дж. У. Гиббсом В (1874).изотермическом равновесном процессе, происходящем при пост. давлении, убыль Г. э. системы равна полной работе системы за вычетом работы против внеш. давления (т. е. равна макс. полезной работе). Г. э. выражают обычно в кДж/моль или кДж/кг.
Если процесс протекает самопроизвольно, то внутренняя энергия (энтальпия) должны уменьшаться, а энтропия увеличиваться. Для сравнения этих величин их надо выразить в одних единицах, а для этого Δ S умножить на T. В этом случае имеем Δ Н – энтальпийный фактор и Т Δ S - энтропийный фактор.
В ходе реакции частицы стремятся к объединению, что ведет к уменьшению энтальпии (Δ Н < 0), с другой стороны – должна возрастать энтропия, т.е. увеличиваться число частиц в системе (Т Δ S > 0). "Движущая сила" реакции определяется разностью между этими величинами и обозначается Δ G.
Δ G p,T = Δ H – T Δ S
и называется изменением энергии Гиббса (изобарно-изотермический потенциал).
Энергия Гиббса - это часть энергетического эффекта реакции, которую можно превратить в работу, поэтому ее называют свободной энергией. Это тоже термодинамическая функция состояния и, следовательно, для реакции
b B + d D = l L + m M,
энергию Гиббса химической реакции можно рассчитать как сумму энергий Гиббса образования продуктов реакции за вычетом энергий Гиббса образования исходных веществ с учетом стехиометрических коэффициентов по формуле:
Δ G = l Δf G L + m Δf G M – d Δf G D – b Δf G B.
где Δf G – энергия Гиббса образования веществ.
Энергия Гиббса образования веществ это изменение энергии Гиббса системы при образовании 1 моль вещества из простых веществ, устойчивых при 298 К.
Энергия Гиббса образования простых веществ Δf G принимается равной нулю. Если образующееся вещество и исходные простые вещества находятся в стандартных состояниях, то энергия Гиббса образования называется стандартной энергией Гиббса образования вещества Δf G 0. Ее значения приводятся в справочниках.
Полученное значение Δ G является критерием самопроизвольного течения реакции в прямом направлении, если Δ G < 0. Химическая реакция не может протекать самопроизвольно в прямом направлении, если энергия Гиббса системы возрастает, т.е. Δ G > 0. Если Δ G = 0, то реакция может протекать как в прямом, так и в обратном направлениях, т.е. реакция обратима.
Направление химических реакций зависит от их характера. Так, условие Δ G < 0 соблюдается при любой температуре для экзотермических реакций (Δ Н < 0), у которых в ходе реакции возрастает число молей газообразных веществ, и, следовательно, энтропия (Δ S > 0). У таких реакций обе движущие силы (Δ Н) и (Т Δ S) направлены в сторону протекания прямой реакции иΔ G < 0 при любых температурах. Такие реакции являются необратимыми.
Наоборот, эндотермическая реакция (Δ Н > 0), в результате которой уменьшается число молей газообразных веществ (Δ S < 0) не могут протекать самопроизвольно в прямом направлении при любой температуре, т.к. всегда Δ G > 0.
Если в результате экзотермической реакции (Δ Н < 0) уменьшается число молей газообразных веществ и, соответственно, энтропия (Δ S < 0), то при невысокой температуре Δ Н > T Δ S и реакция возможна в прямом направлении (Δ G < 0). При высоких температурах Δ H < T Δ S и прямая реакция самопроизвольно протекать не может (Δ G > 0), а обратная реакция возможна.
Для определения температуры равновесия можно воспользоваться условием:
Т р = Δ Н /Δ S,
где Т р – температура, при которой устанавливается равновесие, т.е. возможность протекания прямой и обратной реакций.
Если в результате эндотермической реакции (Δ Н > 0) увеличивается число молей газообразных веществ и энтропия системы (Δ S > 0), то при невысоких температурах, когда Δ Н > Т Δ S, самопроизвольно прямая реакция идти не может (Δ G > 0), а при высоких температурах, когда Δ Н < T Δ S, прямая реакция может протекать самопроизвольно (Δ G < 0).
Связь между Δ G и Δ G 0 выражается уравнением изотермы Вант-Гоффа, которая для реакции
b B + d D = l L + m M
записывается в виде:

либо в виде:
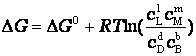
где  - относительные парциальные давления соответствующих веществ;
- относительные парциальные давления соответствующих веществ; 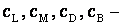 концентрации соответствующих растворенных веществ.
концентрации соответствующих растворенных веществ.
Итак, энергия Гиббса позволяет определить возможность протекания реакции расчетным путем, не прибегая к дорогостоящим и длительным экспериментам.
В изохорно-изотермических условиях свободная энергия называется энергией Гельмгольца или изохорно-изотермическим потенциалом и равна

Она характеризует направление и предел самопроизвольного течения химической реакции при изохорно-изотермических условиях, которое возможно при Δ F < 0.
• 51. Химическая кинетика. Скорость реакции и методы ее регулирования. Способы количе-
ственного выражения и практического определения скорости химической реакции.
ХИМИЧЕСКАЯ КИНЕТИКА – (от греч.кинетикос – движущий) наука о механизмах химических реакций и закономерностях их протекания во времени.
В 19 в. результате развития основ химической термодинамики химики научились рассчитывать состав равновесной смеси для обратимых химических реакций. Кроме того, на основании несложных расчетов можно было, не проводя экспериментов, сделать вывод о принципиальной возможности или невозможности протекания конкретной реакции в данных условиях. Однако «принципиальная возможность» реакции еще не означает, что она пойдет. Например, реакция С + О2 ® СО2 с точки зрения термодинамики весьма благоприятна, во всяком случае, при температурах ниже 1000° С (при более высоких температурах происходит уже распад молекул СО2), т.е. углерод и кислород должны (практически со 100%-ным выходом) превратиться в диоксид углерода. Однако опыт показывает, что кусок угля может годами лежать на воздухе, при свободном доступе кислорода, не претерпевая никаких изменений. То же можно сказать и о множестве других известных реакций. Например, смеси водорода с хлором или с кислородом могут сохраняться очень долго без всяких признаков химических реакций, хотя в обоих случаях реакции термодинамически благоприятны. Это означает, что после достижения равновесия в стехиометрической смеси H2 + Cl2 должен остаться только хлороводород, а в смеси 2Н2 + О2 – только вода. Другой пример: газообразный ацетилен вполне стабилен, хотя реакция C2H2 ® 2C + H2 не только термодинамически разрешена, но и сопровождается значительным выделением энергии. Действительно, при высоких давлениях, ацетилен взрывается, однако в обычных условиях он вполне стабилен.
Термодинамически разрешенные реакции, подобные рассмотренным, могут пойти только в определенных условиях. Например, после поджигания уголь или сера самопроизвольно соединяются с кислородом; водород легко реагирует с хлором при повышении температуры или при действии ультрафиолетового света; смесь водорода с кислородом (гремучий газ) взрывается при поджигании или при внесении катализатора. Почему же для осуществления всех этих реакций необходимы специальные воздействия – нагревание, облучение, действие катализаторов? Химическая термодинамика не дает ответа на этот вопрос – понятие времени в ней отсутствует. В то же время для практических целей очень важно знать, пройдет ли данная реакция за секунду, за год или же за многие тысячелетия.
Опыт показывает, что скорость разных реакций может отличаться очень сильно. Практически мгновенно идут многие реакции в водных растворах. Так, при добавлении избытка кислоты к щелочному раствору фенолфталеина малинового цвета раствор мгновенно обесцвечивается, это означает, что реакция нейтрализации, а также реакция превращения окрашенной формы индикатора в бесцветную идут очень быстро. Значительно медленнее идет реакция окисления водного раствора иодида калия кислородом воздуха: желтая окраска продукта реакции – иода появляется лишь через продолжительное время. Медленно протекают процессы коррозии железных и особенно медных сплавов, многие другие процессы.
Предсказание скорости химической реакции, а также выяснение зависимости этой скорости от условий проведения реакции – одна из важных задач химической кинетики – науки, изучающей закономерности протекания реакций во времени. Не менее важна и вторая задача, стоящая перед химической кинетикой – изучение механизма химических реакций, то есть детального пути превращения исходных веществ в продукты реакции.
Скорость реакции и методы ее регулирования:
Скорость реакции. Проще всего определить скорость для реакции, протекающей между газообразными или жидкими реагентами в гомогенной (однородной) смеси в сосуде постоянного объема. В этом случае скорость реакции определяется как изменение концентрации любого из участвующих в реакции веществ (это может быть исходное вещество или продукт реакции) в единицу времени. Это определение можно записать в виде производной: v = dc/dt, где v – скорость реакции; t – временя, c – концентрация. Эту скорость легко определить, если есть экспериментальные данные по зависимости концентрации вещества от времени. По этим данным можно построить график, который называется кинетической кривой. Скорость реакции в заданной точке кинетической кривой определяется наклоном касательной в этой точке. Определение наклона касательной всегда связано с некоторой ошибкой. Точнее всего определяется начальная скорость реакции, поскольку вначале кинетическая кривая обычно близка к прямой; это облегчает проведение касательной в начальной точке кривой.
Если время измерять в секундах, а концентрацию – в молях на литр, то скорость реакции измеряется в единицах моль/(л·с). Таким образом, скорость реакции не зависит от объема реакционной смеси: при одинаковых условиях она будет одинаковой и в маленькой пробирке, и в многотоннажном реакторе.
Величина dt всегда положительна, тогда как знак dc зависит от того, как изменяется со временем концентрация – уменьшается (для исходных веществ) или увеличивается (для продуктов реакции). Чтобы скорость реакции всегда оставалась величиной положительной, в случае исходных веществ перед производной ставят знак минус: v = –dc/dt. Если реакция идет в газовой фазе, вместо концентрации веществ в уравнении скорости часто используют давление. Если газ близок к идеальному, то давление р связано с концентрацией с простым уравнением: p = cRT.
В ходе реакции разные вещества могут расходоваться и образовываться с разной скоростью, в соответствии с коэффициентами в стехиометрическом уравнении (см. СТЕХИОМЕТРИЯ), поэтому, определяя скорость конкретной реакции, следует учитывать эти коэффициенты. Например, в реакции синтеза аммиака 3H2 + N2 ® 2NH3 водород расходуется в 3 раза быстрее, чем азот, а аммиак накапливается в 2 раза быстрее, чем расходуется азот. Пэтому уравнение скорости для этой реакции записывают следующим образом: v = –1/3 dp(H2)/dt = –dp(N2)/dt = +1/2 dp(NH3)/dt. В общем случае, если реакция стехиометрическая, т.е. протекает точно в соответствии с записанным уравнением: aA + bB ® cC + dD, ее скорость определяют как v = –(1/a)d[A]/dt = –(1/b)d[B]/dt = (1/c)d[C]/dt = (1/d)d[D]/dt (квадратными скобками принято указывать молярную концентрацию веществ). Таким образом, скорости по каждому веществу жестко связаны между собой и, определив экспериментально скорость для любого участника реакции, легко рассчитать ее для любого другого вещества.
Большинство реакций, используемых в промышленности, относятся к гетерогенно-каталитическим. Они протекают на поверхности раздела фаз между твердым катализатором и газовой или жидкой фазой. На поверхности раздела двух фаз протекают и такие реакции как обжиг сульфидов, растворение металлов, оксидов и карбонатов в кислотах, ряд других процессов. Для таких реакций скорость зависит и от величины поверхности раздела, поэтому скорость гетерогенной реакции относят не к единице объема, а к единице поверхности. Измерить величину поверхности, на которой идет реакция, не всегда просто.
Если реакция протекает в замкнутом объеме, то ее скорость в большинстве случаев максимальна в начальный момент времени (когда максимальна концентрация исходных веществ), а затем, по мере превращения исходных реагентов в продукты и, соответственно, снижения их концентрации, скорость реакции уменьшается. Встречаются и реакции, в которых скорость увеличивается со временем. Например, если медную пластинку опустить в раствор чистой азотной кислоты, то скорость реакции будет расти со временем, что легко наблюдать визуально. Ускоряются со временем также процессы растворения алюминия в растворах щелочей, окисления многих органических соединений кислородом, ряд других процессов. Причины такого ускорения могут быть разными. Например, это может быть связано с удалением защитной оксидной пленки с поверхности металла, или с постепенным разогревом реакционной смеси, или с накоплением веществ, ускоряющих реакцию (такие реакции называются автокаталитическими).
В промышленности реакции обычно проводят путем непрерывной подачи в реактор исходных веществ и вывода продуктов. В таких условиях можно добиться постоянной скорости протекания химической реакции. С постоянной скоростью протекают и фотохимические реакции при условии полного поглощения падающего света (см. ФОТОХИМИЧЕСКИЕ РЕАКЦИИ).
Лимитирующая стадия реакции. Если реакция осуществляется путем последовательно протекающих стадий (не обязательно все из них являются химическими) и одна из этих стадий требует значительно большего времени, чем остальные, то есть идет намного медленнее, то такая стадия называется лимитирующей. Именно эта самая медленная стадия определяет скорость всего процесса. Рассмотрим в качестве примера каталитическую реакцию окисления аммиака. Здесь возможны два предельных случая.
1. Поступление молекул реагентов – аммиака и кислорода к поверхности катализатора (физический процесс) происходит значительно медленнее, чем сама каталитическая реакция на поверхности. Тогда для повышения скорости образования целевого продукта – оксида азота совершенно бесполезно повышать эффективность катализатора, а надо позаботиться об ускорении доступа реагентов к поверхности.
2. Подача реагентов к поверхности происходит значительно быстрее самой химической реакции. Вот здесь имеет смысл совершенствовать катализатор, подбирать оптимальные условия для каталитической реакции, так как лимитирующей стадией в данном случае является каталитическая реакция на поверхности.
• 52. Влияние различных параметров на скорость химической реакции. Основное кинетическое
уравнение (закон действующих масс). Константа скорости. Правило Вант-Гоффа и уравне-
ние Аррениуса.
Скорость химической реакции зависит от природы реагирующих веществ и условий протекания реакции: концентрации с, температуры t, присутствия катализаторов, а также от некоторых других факторов (например, от давления - для газовых реакций, от измельчения - для твердых веществ, от радиоактивного облучения).
Влияние концентраций реагирующих веществ. Чтобы осуществлялось химическое взаимодействие веществ А и В, их молекулы (частицы) должны столкнуться. Чем больше столкновений, тем быстрее протекает реакция. Число же столкновений тем больше, чем выше концентрация реагирующих веществ. Отсюда на основе обширного экспериментального материала сформулирован основной закон химической кинетики, устанавливающий зависимость скорости реакции от концентрации реагирующих веществ:
Cкорость химической реакции пропорциональна произведению концентраций реагирующих веществ.
Для реакции (I) этот закон выразится уравнением
v = kcA cB, (1)
где сА и сВ - концентрации веществ А и В, моль/л; k - коэффициент пропорциональности, называемый константой скорости реакции. Основной закон химической кинетики часто называют законом действующих масс.
Из уравнения (1) нетрудно установить физический смысл константы скорости k: она численно равна скорости реакции, когда концентрации каждого из реагирующих веществ составляют 1 моль/л или когда их произведение равно единице.

