 2015-05-13
2015-05-13 6082
6082Существует несколько механизмов ориентирования макромолекул. В природе рост целлюлозных (хлопок, лён, конопля, джут и др.) и белковых (шерсть) волокон определяет продольную ориентацию макромолекл, образующих фибриллярную структуру. В искусственных и синтетических полимерах ориентация макромолекул определяется технологическими приёмами переработки. Наиболее полно ориентационные эффекты проявляются в плёнках и, особенно, в волокнах. Технология изготовления этих изделий, как правило, предполагает выдавливание (экструзию) раствора или расплава полимера через калиброванные отверстие определённой формы – фильеру. В зависимости от метода формования (расплав, мокрое формование раствора, сухо-мокрое формование раствора, сухое формование раствора) ориентационные процессы протекают по-разному. Важно учитывать, что все структурные изменения в полимерах протекают во времени, являются релаксационными. Это проявляется, например, при фазовых переходах в покоящейся системе при разных скоростях изменения температуры или в структурных переходах под действием внешнего механического поля. Наиболее отчётливо роль структурно-релаксационных факторов проявляется в процессах «ориентационного структурообразования», т.е. при продольном течении растворов и расплавов кристаллизующихся и аморфных полимеров, неизотермической кристаллизации во внешнем механическом поле и, в особенности, в процессе превращения «струя – волокно». Уже на стадии формования при продольном течение через отверстия фильеры происходит ориентация макромолекул вдоль оси волокна. Молекулярные модели полимерных жидкостей при ориентировании в струе представлены на рис. 18.

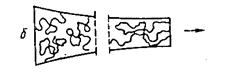

Рис.18. Молекулярные модели полимерных жидкостей при ориентировании в струе:
а – жёсткие эллипсоиды; б – разбавленный раствор гибких цепных макромолекул;
в – подвижная сетка с локальными диссоциирующими узлами.
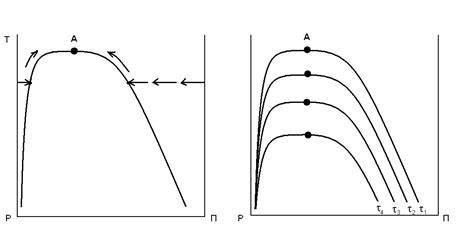
Однако, для гибкоцепных полимеров ориентация, достигаемая на этой стадии, недостаточна, тем более, что в области выхода из фильеры релаксация протекает настолько быстро, что практически сводит на нет достигнутый в канале фильеры эффект. Основная ориентация создаётся на стадии упрочняющей вытяжки, величина которой тем больше, чем выше кратность вытягивания. А кратность вытягивания зависит от температуры, продолжительности (скорости вытягивания) и усилия вытяжки. Процесс вытягивания характеризуется кривыми растяжения в изотермических условиях (кривые σ-ε). На рис. 19 приведены кривые растяжения полиамидных волокон при разных температурах. (М.П.Носов в «Теории формования химических волокон» М. Химия, 1975 г с. 178)
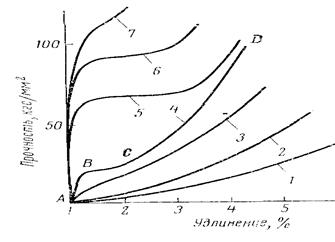
Рис. 19. Кривые растяжения полиамидных волокон при различных температурах (в оС). 7 – -200; 6 – -170; 5 – -100; 4 – - 20; 3 - -15; 2 - +50; 1 - +75. По оси абсцисс – не прочность, а усилие (напряжение) вытягивания.
Максимальная вытяжка, выше которой прочность начинает уменьшаться, для разных полимеров достигается при разных, присущих данному полимеру, температурах. Иначе говоря, создаётся впечатление, что для каждого полимера существует технологический предел кратности вытягивания и соответственно – достижимой величины прочности. Это ограничение побуждает разрабатывать специальные технологические приёмы повышения кратности вытягивания. Так, в 1964 г Бондаренко В.М., Бычковым Р.А. и Зверевым М.П. был предложен «Способ одноступенчатой вытяжки синтетических волокон», позволяющий увеличить кратность вытягивания и соответственно - прочность путём вытяжки выше температуры плавления полимера (А.с. № 361234), что достигалось вытягиванием на специальных градиентных нагревателях, обеспечивающих постепенное повышение температуры (А.с. № 347377). А в 80-х годах была разработана «гель-технология», позволившая достичь рекордных прочностей (см. раздел 3)..
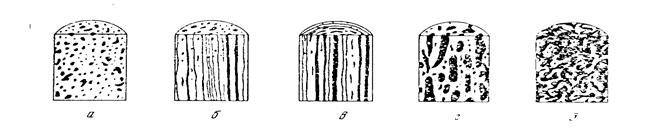
В кристаллических полимерах при вытягивании происходит разрушение («плавление») кристаллов по одному из возможных механизмов с образование фибриллярной структуры путём распрямления макромолекул из нескольких ламелей (Кабояши) и путём постепенного наклонения цепей, их скольжения друг по другу и распада кристалла на отдельные блоки из складчатых цепей (Петерлин). Схемы разрушения кристаллов по Кабояши (а) и Петерлину (б) представлены на рис. 20.
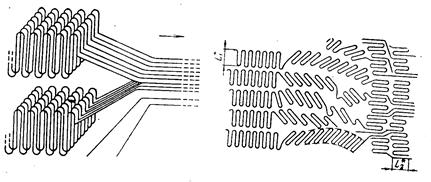
Рис.20. Молекулярный механизм пластической деформации полимерных кристаллов: а) по Кобаяши: б) по Петерлину.
Структура, образующаяся после деформирования (вытягивания), подвергается дальнейшим изменениям при термической обработке или действии растворителей. Со структурной точки зрения здесь происходит ориентационная вторичная кристаллизация. Размеры кристаллитов при этом растут, их упорядоченность увеличивается. Однако такая обработка может несколько уменьшить молекулярную ориентацию.
В жёсткоцепных полимерах, формуемых из раствора, молекулярная ориентация может быть заложена ещё на стадии приготовления раствора. Дело в том, что некоторые растворы жёсткоцепных полимеров могут находиться в жидкокристаллическом состоянии (в мезофазе). В отличие от низкомолекулярных жидких кристаллов мезогенность полимерной молекулы определяется длиной не всей цепочки, а величиной статистического сегмента, длина которого составляет несколько сотен ангстрем.
Установлено, что переход жёсткоцепных полимеров при растворении в упорядоченное состояние носит характер фазового перехода первого рода, при котором возникает структура нематического типа. По мере увеличения концентрации вязкость раствора возрастает, а при достижении критической концентрации – резко падает. При больших напряжениях сдвига (перемешивание) максимум не проявляется и растворы ведут себя, как обычные изотропные. Из этого следует, что интенсивное перемешивание препятствует образованию жидкокристаллической структуры.
Переход из изотропного состояния в анизотропное регистрируется:
а) поляризационно-оптическими и визуальными наблюдениями. Растворы становятся мутными, иногда – опалесцирующими;
б) реологическими методами. Максимум вязкости соответствует точке обращения изотропной матрицы с включениями областей анизотропных образований в анизотропную матрицу с включением изотропных областей. Волокна, сформованные из анизотропных растворов, более прочны, чем сформованные из изотропных растворов. Кристалличность волокон из анизотропных растворов выше, чем из изотропных.
При течении мезофазного раствора через канал фильеры происходит ориентация нематических кристаллов вдоль оси волокна, что непосредственно обеспечивает значения прочности 2 – 4 ГПа. Основным видом надмолекулярных образований в этих волокнах являются фибриллы. Микрофибрилла состоит из кристаллитов и аморфных прослоек. Молекулярные цепи в аморфных прослойках почти параллельны осям кристаллитов. Микрофибриллы располагаются вдоль оси волокна (часть из них – под углом 100)
Не все термостойкие жёсткоцепные сверхпрочные волокна кристалличны. Так, волокно «вниивлон» - аморфно. Общее у всех сверхпрочных волокон – число молекулярных цепей, приходящихся на единицу поперечного сечения волокна. Число проходных цепей, держащих нагрузку, в нагруженном образце составляет не менее 0,75, а фактор ориентации – не менее 0,95.
«Жёсткость» можно искусственно придать гибкоцепным полимерам энергией внешнего поля, т.е. растянуть макромолекулы гидродинамическим или механическим полем, при этом также может возникнуть нематическая структура.
В общем случае прочность материалов зависит от типа химических связей между атомами материала, от структуры материала. Различают теоретическую, реальную и эксплуатационную прочность, т.е. ту, которую закладывают в расчёты конструкций. Теоретическую прочность рассчитывают исходя из величин сил межатомного взаимодействия, имеющих место в данном материале. Для полимеров расчёт теоретической прочности основывается на оценке работы разрыва макромолекул по линии основных связей полимерных цепей. Предполагается, что в идеальном случае макромолекулы плотно упакованы и расположены своими осями строго вдоль направления растягивающего усилия. При этом длина молекул рассматривается как бесконечно большая, т.е. разрыв происходит одновременно всех макромолекул, приходящихся на поперечное сечение образца. Расчёты показывают, что при температуре абсолютного нуля прочности близки к 6 – 8 ГПа.
С ростом температуры прочность падает и при 250С (2980К) составляет 0,55 – 0,65 от теоретической. Значения реальной прочности многих полимеров достигли, а порой и превосходят теоретические расчёты. Вероятно, это связано с вкладом сил межмолекулярного взаимодействия, не учитываемых в теоретическом расчёте. Понятно, что наибольшие прочности достигаются для ориентированных материалов, когда в сечение разрыва попадает как можно больше связей. Мерой ориентации принят угол разориентации a - угол между осью волокна и средним углом ориентации макромолекул. На рис. 21 представлена зависимость относительной прочности s /sтеор. от угла разориентации a. Как видно из рис. 21, при уменьшении угла разориентации до 300, т.е. до состояния, которому отвечают волокна, вышедшие из осадительной ванны и не подвергнутые дополнительной высокой ориентационной вытяжке, относительная прочность составляет менее 0,1 от максимальной прочности идеально ориентированного волокна. Затем, по мере уменьшения угла разориентации, прочность начинает резко увеличиваться. При угле разориентации 80 прочность составляет половину максимальной прочности, т.е. 2500 МПа. В действительности при такой ориентации достигается прочность не выше 1000 МПа. Причиной такого расхождения является дефектность макро- и микроструктуры волокон. Она, в свою очередь, зависит от условий формования, определяющих кинетику и механизм разделения фаз при нарушении равновесия в системах полимер – растворитель.
 s / s0
s / s0
 1 -
1 -
0,5 Рис.21. Зависимость относительной
прочности от угла разориентации.
 |
0 10 20 30 40 Угол разориентации a.
Решающее влияние на реальную (физическую) прочность оказывают поверхностные дефекты в виде микротрещин. Эту гипотезу вначале 20-х годов прошлого века высказал Гриффит. Согласно его представлениям разрушение твёрдых тел происходит мгновенно после достижения некоторого критического (предельного) напряжения. При нагружении твёрдого тела в вершинах микротрещин возникают большие местные перенаряжения, превышающие средние напряжения в десятки и даже сотни раз. Когда эти перенапряжения превысят силы взаимодействия между соседними структурными элементами, трещина начинает расти с большой скоростью и твёрдое тело разрушается. Именно эти микротрещины понижают прочность по сравнению с теоретической в сотни раз. Впервые это было экспериментально доказано исследованиями, проводившимися в Физико-техническом институте Академии наук под руководством А. Ф. Иоффе на примере кристаллов каменной соли. Теоретическая прочность на растяжение кристаллов NaCl приблизительно составляет 2 ГПа, а реально при комнатной температуре прочность не превышает 5 МПа, т.е., в 400 раз ниже. В то же время при испытаниях, проводившихся в горячей воде, прочность повышалась в десятки раз и, в ряде случаев, приближалась к теоретической. Горячая вода растворяла дефектный поверхностный слой, содержащий трещины (концентраторы напряжения), и прикладываемое напряжение при этом распределяется на всё поперечное сечение.
Эти закономерности чётко прослеживаются на материалах любого типа. Так, нитевидные кристаллы железа, представляющие собой практически монокристалл с малым количеством дислокаций, имеют предел прочности при растяжении порядка 10 ГПа, а поликристаллическое железо, очищенное методом зонной плавки, имеет предел прочности при растяжении всего 0,1 ГПа. Легированные стали после специальной термообработки обладают прочность порядка 2 – 3 ГПа. А современные высокопрочные волокна имеют сейчас прочность выше 6 ГПа. Их структура закладывается на стадии формования из раствора полимера.
3. Фазовое равновесие в системе полимер – растворитель.
(здесь, как и везде в этом пособии, под термином «растворитель» понимается низкомолекулярная жидкость, взаимодействующая с полимером, но не обязательно переводящая его в раствор).
Все системы полимер – растворитель можно разделить на следующие основные группы:
1) истинные растворы с преобладающим содержанием растворителя;
2) пластифицированные полимеры;
3) студни;
4) двухфазные системы с полным разделением фаз;
5) дисперсии полимеров.
Для конденсированных многокомпонентных систем наблюдается два основных вида фазового равновесия – аморфное и кристаллическое. В области ограниченной совместимости компонентов система распадается на две фазы. В случае аморфного равновесия обе фазы представляют собой насыщенные растворы одного компонента в другом. При кристаллическом равновесии одна фаза представляет собой кристаллический осадок (в предельном случае – монокристаллы) одного из компонентов, а вторая фаза – насыщенный раствор этого компонента в другом компоненте.
Аморфное равновесия в системе полимер – растворитель.
Всегда кривая фазового равновесия для системы полимер – растворитель сдвинута в сторону малых концентрация полимера (больших концентраций растворителя), рис. 22.
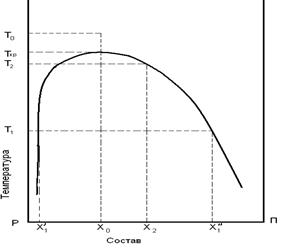
Рис. 22. Фазовая диаграмма аморфного равновесия системы полимер – растворитель.
Выше кривой (её называют бинодалью) система представляет собой раствор полимера в растворителе.
При охлаждении ниже температуры t1 раствор полимера распадается на две фазы – фазу, богатую растворителем х1 и фазу, богатую (обогащённую) полимером х2. Если фаза х1 представляет собой почти чистый растворитель, то фаза х2 в зависимости от концентрации в ней полимера, может представлять собой раствор полимера в растворителе, студень или рыхлый осадок. В начальной стадии перехода системы от однофазного раствора к двухфазному состоянию должны появляться зародыши обеих фаз. Зародыши фазы 1 образуются быстрее, т.к. скорость диффузии низкомолекулярных молекул растворителя из однофазного неравновесного раствора выше, чем скорость диффузии макромолекул полимера. В этом случае фаза 2 (фаза, богатая полимером) образует непрерывный коагуляционный каркас, в ячейках которого заключена фаза 1. Такая структура называется студнем. Именно такой структурой обладают волокна, формуемые из растворов полимеров.
Если же скорость образования зародышей фазы 2 больше, чем фазы 1, то образуются рыхлые осадки фазы 2 – фазы, богатой полимером.
При формовании волокон, плёнок и покрытий из растворов полимеров обычно имеют дело с аморфным равновесием в системе полимер – растворитель и образованием структуры студня. Именно на этом направлении в 80-е годы ХХ столетия были достигнуты выдающиеся успехи – была разработана так называемая гель-технология.
Сущность процесса получения полиолефиновых, в частности, полиэтиленовых волокон по гель-технологии заключается в том, что волокна формуют из горячего раствора полимера в холодную водную осадительную ванну (осаждающую жидкость), которая не смешивается с растворителем, а лишь играет роль эффективного охладителя. В результате переохлаждения в струе прядильного раствора наступает фазовое разделение системы и образуется гель (студень), содержащий исходный растворитель, который при этой температуре уже не обладает растворяющей способностью к полимеру. Гель-волокно сушится либо другим путём (отмывается) освобождается от растворителя, ячеистая структура студня при этом сохраняется. Отмытое или высушенное волокно, называемое «ксерогелем» (твёрдым гелем), имеет хорошую способность к ориентационному вытягиванию и повышению механических характеристик. В специально подобранных условиях удалось достичь прочности 6 – 7 ГПа, а модуль упругости» 200 ГПа. Считается, что именно стадия образования геля обеспечивает возможность последующей многократной вытяжки и резкого повышения механических свойств полиэтиленовых волокон. Ещё одним обязательным условием является использование сверхвысокомолекулярного полимера с молекулярной массой = 5 . 105 – 8 . 106 .
Вернёмся к рассмотрению фазовой диаграммы. Кривая фазового равновесия практически представляет собой не одну линию (бинодаль), а систему кривых, отвечающих ММР (молекулярно-массовому распределению). Эта область толще в левой ветви кривой фазового равновесия, как показано на рис. 23. Нижнюю ограничивающую кривую называют спинодалью.
Рис.23. (Либо рис. 14 из Ф-Х основ переработки…, либо рис. 111.2 из Студенобразн сост.)
Как известно, бинодаль представляет собой границу устойчивости гомогенной фазы, а спинодаль характеризует границу абсолютной неустойчивости гомогенной фазы.
Области, заключённые между бинодальной и спинодальной кривыми и под спинодальной кривой, соответствуют различной степени стабильности неравновесного раствора. Соответственно, могут проявляться различные механизмы распада неравновесного раствора на фазы. Между бинодалью и спинодалью система находится в метастабильном состоянии, т.е., она устойчива к небольшим флуктуациям концентрации. Если же флуктуации превышают определённый критический размер, возникают зародыши новой фазы, как описано выше.
Наличие области между бинодалью и спинодалью позволяет производить фракционирование полимеров по Мм путём ступенчатого охлаждения раствора и отфильтровывания образующихся осадков.
Механизм распада неравновесной системы на две фазы по схеме образования зародышей и их роста до установления полного равновесия между фазами по составу применим к любому случаю превращений с образованием новых фаз: к выделению газов из пересыщенных растворов, к кристаллизации веществ из пара, из жидкого раствора, из расплава и из твёрдого аморфного вещества.
Если же система попадает в область, лежащую внутри спинодали, то может реализоваться другой механизм распада, называемый спинодальным распадом. Здесь возможно образование больших областей, очень мало отличающихся по составу от неравновесного исходного раствора. В результате может образоваться система непрерывных фаз, которые не являются равновесными по составу. Если температура относительно высока, то текучесть полимерной фазы оказывается достаточной для того, чтобы произошло нарушение её непрерывности и превращение в сферические образования. Образование студня при спинодальном распаде маловероятен.
Существует другой механизм образования студней – путём набухания специальным образом приготовленного порошкообразного полимера (пастообразуюшего полимера) в пластификаторе. Такие системы называются полимерными пластизолями. При смешивании пастообразующего полимера с пластификатором протекает ряд физико-химических процессов: смачивание, пропитка полимерных агломератов пластификатором, развал агломератов на отдельные частицы, набухание частиц.
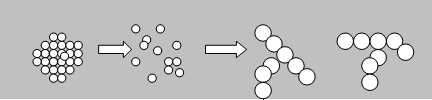
Рис. 24. Стадии структурообразования в процессе старения пластизолей.
При определённой степени набухания частицы будут соприкасаться друг с другом, образуя коагуляционные структуры различной сложности, вплоть до пространственных сотоподобных структур, т.е. студней.
Рис.25. Возможная структура студня на основе пастообразующего полимера в изображении на плоскости.
В этих студнях стенки ячеек образованы набухшими и продолжающими набухать частицами полимера (фаза, богатая полимером), а внутри ячеек находится низкоконцентрированный раствор полимера в пластификаторе. Конечным состоянием такой системы является состояние пластифицированного полимера – истинного раствора пластификатор в полимере. В таком состоянии материал обладает свойствами эластомера. Термодинамика образования студней на основе пластизолей пока окончательно не разработана. Есть представления (спорные), развиваемые на кафедре ТИ-6 Бычковым Р.А. [ ].
В случае пластизолей система изначально двухфазна: исходный полимер П и чистый растворитель (пластификатор) Р – рисунок 26.
t4 > t3 >t2 > t1
Рис. 26. Гипотетическое изменение фазовой диаграммы системы пастообразующий полимер – пластификатор во времени.
Состав этих фаз по мере протекания процессов смачивания, пропитки и набухания полимерных частиц изменяется от П и Р до П1 и Р1, находящихся на бинодали. При достижении бинодали пластизоль превращается в студень (пластигель). Однако на этом превращения в такой системе не прекращаются: - набухание полимерных частиц продолжается. Это равносильно восхождение состава фаз П1 и Р1 по бинодали вверх до точки А. Но, поскольку эксперимент проводится в изотермических условиях, следует предположить, что бинодаль тонет, погружается в область более низких температур. Это приводит к уменьшению объёмной доли фазы, богатой растворителем и увеличению объёмной доли фазы, богатой полимером. В точке А система превращается в истинный раствор пластификатора в полимере – в пластифицированный полимер. В термодинамике такие движения бинодали не рассматриваются. Возможно, если воспользоваться эквивалентностью влияния температуры и пластификатора на время релаксации полимеров, удастся оправдать восхождение состава фаз по бинодали вверх до точки А или погружение бинодали. Следует отметить, что для разных полимерных пластизолей эти изменения, приводящие в итоге к образованию однофазной системы полимер – пластификатор (точка А), протекают с разной скоростью. Существуют пластизоли, в которых завершение изменений происходит от нескольких минут до нескольких часов (в зависимости от температуры термостатирования), а в пластизолях ПВХ, напротив, при обычных температурах не завершается и за несколько лет.
Фазовое равновесие в системе кристаллизующийся полимер – низкомолекулярная жидкость.
По сравнению с двухкомпонентной системой кристаллизующихся низкомолекулярных веществ, не образующих твёрдых растворов, диаграмма состояния системы кристаллизующийся полимер – растворитель имеет существенные и принципиальные различия. Сравним эти системы.

 Т
Т


 Тв Т
Тв Т

 Т1 --------------------
Т1 --------------------
 Та 1
Та 1

 2 3
2 3
Тэ -----------------------
 |  |
100%А хэ х1 100%В Р Состав П
Рис. 23. Диаграмма состояния Рис. 24. Сдвиг эвтектической точки
двухкомпонентной системы низко- для системы кристаллизующийся молекулярных кристаллизующихся полимер - растворитель.
веществ..
В низкомолекулярных кристаллизующихся веществах (рис. 23) область 1 соответствует однофазному жидкому раствору компонентов А и В. При температуре Т1, находящейся выше температуры плавления кристаллов А, но ниже температуры плавления кристаллов В, начиная от 100% А и вплоть до состава х1, система однофазна. При более высокой концентрации вещества В (при той же температуре Т1) начинается частичная кристаллизация его, и в области 3 система уже двухфазна: насыщенный раствор В в А и кристаллы В. Соответствующие явления наблюдаются и в области 2, где выделяются кристаллы А. Кривые температур плавления системы (линии ликвидуса) выпуклы по отношению к оси состава и пересекаются в точке эвтектики, отвечающей составу хэ.
В системе полимер – низкомолекулярный растворитель (рис.24) точка эвтектики резко сдвигается по оси состава в сторону растворителя. Различие между температурой кристаллизации чистого растворителя и температурой в точке эвтектики ничтожно. В относительно концентрированных растворах полимеров (тех, что используются в технике) можно вообще игнорировать наличие области эвтектических составов и кривую ликвидуса рассматривать как монотонно изменяющуюся во всей области составов. Так, для системы полиэтилен – ксилол эвтектическая точка соответствует концентрации полиэтилена менее 0,01%. Можно принять для практических целей, что фаза раствора над кристаллизующимся полимером представляет собой чистый растворитель.
Полимерные кристаллы, в том числе и монокристаллы, дефектны, степень несовершенства даже для полиэтилена достигает 15 – 30%. Кристаллическая фаза представляет собой сочетание кристаллических и аморфных областей, поэтому структура полимерных кристаллов является аморфно-кристаллической. Скорость установления равновесия в полимерных системах мала.
3.1. Фазовая структура в смесях полимеров.
Структура смесей полимеров, так же, как и структура любых многокомпонентных систем в твёрдом состоянии, может представлять собой либо механическую смесь, в которой компоненты образуют свои собственные фазы в виде конгломератов макроскопических зерен, либо твердый раствор, когда компоненты диспергированы до уровня атомов, ионов или молекул и образуют общую кристаллическую решетку со статистическим (случайным) расположением атомов в узлах решетки. Такие кристаллы еще называют «изоморфнозамещенными» или фазами переменного состава.
Возможно также химическое взаимодействие между компонентами. В металлических сплавах в этом случае образуются так называемые «интерметаллические соединения».
И, конечно, возможно различное сочетание перечисленных выше структур, например, одновременное присутствие фаз исходных компонентов (механическая смесь) и фаз, отвечающих их твердым растворам.
Однако в смесях полимеров имеется целый ряд особенностей и отличий, обусловленных их строением, а именно:
а) огромными молекулярными массами и, соответственно, огромными размерами макромолекул, ММР;
б) цепным строением макромолекул и, соответственно, анизотропией их размеров;
в) гибкостью макромолекул и, соответственно, конформационным набором «поворотных изомеров»;
г) стереоизомерией.
В результате, в противоположность металлам, полимеры чаще имеют аморфное строение, а кристаллизующиеся полимеры обладают аморфно-кристаллической структурой, т.е. присутствием одновременно и аморфной и кристаллической фаз, причем одна и та же макромолекула может выходить из кристалла в аморфную область и затем входить в другой кристалл.
Условиями, определяющими изоморфное замещение в смесях неорганических веществ являются: 1) близость размеров замещающих друг друга атомов или ионов; 2) близость размеров и формы элементарных ячеек; 3) сохранение типа химической связи в кристалле; 4) совпадение знаков зарядов замещающих друг друга компонентов. Различия в размерах не должны превышать 15%.
Чем сложнее строение частиц, из которых образуется кристалл, тем жёстче требования к их геометрическому подобию. Для полимеров допустимые различия в поперечных размерах макромолекул не превышают 1%. Отсюда понятно, что твёрдые растворы полимеров – чрезвычайно редкое явление.
Структуру смесей кристаллизующихся полимеров можно изучать прямыми структурными методами – по температурам фазовых переходов, рентгеноструктурными методами и др.
Структуру же смесей аморфных полимеров изучать довольно сложно, т.к. практически для этого случая нет прямых структурных методов.
Большое практическое значение имеют растворы смесей полимеров, например, клеи. Однако конечное состояние клея в изделии – твердое агрегатное, т.е. полимерный сплав (блок).
Полимерные системы, в состав которых входят два полимера, начали применять давно для получения материалов, сочетающих свойства обоих смешиваемых полимеров. Еще в начале прошлого столетия хрупкость полистирола уменьшали добавлением каучука. Это привело к созданию широкой гаммы ударопрочных пластмасс, хрупкость которых удалось резко понизить благодаря наличию микрофазы эластомера. Содержание эластомера обычно не превышает 10 – 15 %. При б о льшем количестве уменьшается упругие свойства композиции. Эффект повышения ударной вязкости наблюдается в области температур от 10 до 70 оС. По сравнению с сополимерами смеси гомополимеров того же состава (например, сополимер стирола с 30% акрилонитрила и смесь 70% полистирола и 30% полиакрилонитрила) обладает меньшей прочностью, но зато она способна к большим вынужденно-эластическим деформациям. Это приводит к увеличению энергии разрыва приблизительно в 100 раз по сравнении с сополимером.
Подбирая соответствующие компоненты, можно увеличить модуль упругости, понизить ползучесть (полипропилен – полистирол), повысить прочность, изменить поверхностные свойства, например, придать волокнам способность окрашиваться (полипропилен – поливинилпиридин) и т.д.
Полимерные крспозиции можно приготавливать либо смешением в расплаве, либо в растворе в общем растворителе. Смешение через расплав, естественно, осуществляют при температуре выше их температуры текучести,. при этом полимеры находятся в вязкотекучем (аморфном) состоянии независимо от того, были полимеры до нагревания кристаллическими или аморфными стеклообразными. Таким образом, процесс смешения полимеров в большинстве случаев является процессом взаимодиспергирования и, возможно, взаимного растворения двух или более вязкоупругих жидкостей.
Термодинамика растворения полимера в полимере – это термодинамика взаиморастворения жидких фаз, особенностью которых является участие в процессе растворения гибких молекул достаточно большой длины.
Смешение жидкостей протекает самопроизвольно вплоть до образования истинного раствора (однофазной системы), если процесс сопровождается уменьшением энергии Гиббса, т.е. DG = DH - TDS < 0. Это возможно, если 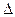 Нm < 0 (выделение тепла) или, даже при Hm> 0, если H < T S.
Нm < 0 (выделение тепла) или, даже при Hm> 0, если H < T S.
Существует мнение, что S полимеров мало, им можно пренебречь и всё определяется изменением ΔН. Так, T S идеальной смеси низкомолекулярных жидкостей 1 см3 + 1 см3 = 34,8 Дж/2см3, TDS 1см3 полимера с М=105 + 1см3 НМ жидкости = 18,9 Дж/2см3, T S 1см3 полимера + 1 см3 полимера = 0,0348 Дж/2см3. Однако, как показывает практика, только по знаку DН определить совместимость полимеров невозможно.
Кстати, как устанавливали совместимость полимеров:
а) смешивают равноконцентрированные растворы двух полимеров в общем растворителе. Отсутствие расслаивания принимается за указание на взаимную растворимость полимеров. Это неверно, т.к. концентрация полимера в растворе невелика (<10 - 15%) и неизвестно что будет, когда растворитель удалят (мутная пленка из прозрачного раствора).
б) если пленка получилась прозрачная – это еще не доказывает взаимного растворения (совместимости), т.к. во-первых, в пленках в отсутствие растворителя равновесие практически не достигается (что будет со временем?) и, во-вторых, если частицы выделившейся фазы £ 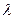 (длина волны света), то пленка будет казаться прозрачной.
(длина волны света), то пленка будет казаться прозрачной.
в) смешивают полимеры на вальцах или другом смесительном оборудовании (экструдер) в отсутствии растворителя. Полученные смеси очевидно неравновесны, т.к. процесс взаиморастворения вязкоупругих жидкостей может не завершится или, наоборот, интенсивное перемешивание, сопровождающееся механохимическими реакциями, может привести к получению однофазной смеси несовместимых полимеров вследствие, механодеструкции и последующей прививки. Так, например, смешение несовместимых полимеров в условиях сдвига под очень большим давлением (наковальня Бриджмена) приводит к образованию однофазной системы. Однако, отжиг при температурах выше температуры стеклования с целью ускорения релаксационных процессов вызывает разделение однофазной системы на исходные полимерные фазы. Кстати, эта система в однофазном состоянии обладает меньшей стойкостью к термодеструкции, чем в двухфазном состоянии.
Очень небольшое число смесей можно надежно отнести к числу взаиморастворимых во всем диапазоне концентраций. Это смесь нитроцеллюлозы (НЦ) и поливинилацетата (ПВА), однофазность которой при получении из разных растворителей была подтверждена разными методами. Эти полимеры во всей области составов смешивались экзотермически ( Hm< 0), при этом во всей области составов G < 0. Однофазна смесь цис-полиизопрена (НК) и разветвленного полибутадиена СКБ. Можно получить однофазные смеси ПВХ и СКН-40. Некоторые полимеры образует однофазные смеси в узком диапазоне составов, обычно не более 5%.
Следует отметить, что определение термодинамических параметров ( H и S) смесей полимеров – трудоёмкое и кропотливое занятие. И чаще всего это делается в системе полимер 1 – полимер 2 – растворитель.
Из кристаллических полимеров твёрдые растворы (изоморфизм замещения) образовывали поливинилфторид с поливинилиденфторидом, поливинилиденфторид с некоторыми сополимерами тетрафторэтилена и винилиденфторида, сополимеры тетрафторэтилена и винилиденфторида близких составов.
3.2. Истинные и коллоидные растворы смесей полимеров.
Известно, что в растворах неполярных полимеров при очень малой концентрации макромолекулы разобщены растворителем настолько, что находятся в виде отдельных клубков. При увеличении концентрации клубки контактируют друг с другом. Эту концентрацию можно определить по перегибу на кривой состав – свойство, например, по светорассеянию. Если для системы П1 –Р положение этой точки зависит главным образом от молекулярной массы М полимера, то для системы П1 - П2 – Р положение точки может отвечать либо критической концентрации одного из полимеров П, либо смесью П1-П2 в целом. Разработаны методы, разрешающие эти сомнения.
Двухфазный мутный раствор смеси полимеров с течением времени может разделиться на два слоя, различающихся по химическому составу и концентрации полимеров в слоях. Чем выше концентрация раствора смеси полимеров, при которой он расслаивается, тем шире концентрационная область существования однофазных, термодинамически устойчивых систем.
На предел расслаивания раствора смеси полимеров влияют следующие факторы: (Продумать, может это усилить)
· Природа и структура макромолекул смешиваемых полимеров;
· Соотношение полимеров в смеси;
· Молекулярная масса М и молекулярно-массовое распределение ММР;
· Химическая природа растворителя, его сродство к полимерам смеси;
· Температура;
· Напряжение сдвига (если раствор деформируется – течет или перемешивается);
· Малые добавки других веществ.
Природа и структура смешиваемых полимеров.
Если полимеры химически не взаимодействуют друг с другом, то, понятно, что чем ближе полимеры по химической природе, тем выше концентрация раствора, при которой полимеры расслаиваются.
Если различие в химической природе велико, то расслаивание может произойти при концентрации менее 1%.
Сильное различие в химической природе, например, наличие полярных функциональных групп, способных взаимодействовать друг с другом, может, наоборот, привести к образованию смесей, не расслаивающихся в растворе.
СКМВП – СКДКТР
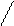
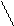
метилвинилпиридин карбоксильные группы
Однако, высказанные выше соображения о важности близости химической природы, не являются абсолютными. Так, растворы поли-о-метилстирола и поли-n-метилстирола (изомеры) расслаиваются.
[-СН2-СН-]n [-СН2-СН-]n




 СН3 ½
СН3 ½

СН3
поли-о-метилстирол поли-n-метилстирол
Более того, расслаиваются растворы натурального каучука (НК) и гуттаперчи, которые являются стереоизомерами (НК – цис-полиизопрен, гуттаперча – транс-полиизопрен).
Это указывает на важность структурного фактора, который, как указывалось ранее, является определяющим при образовании совместных кристаллов в смеси кристаллических полимеров.
Молекулярная масса М и молекулярно-массовое распределение ММР.
С возрастанием молекулярной массы (М) взаимная растворимость полимеров стремится к нулю даже в общем растворителе и наоборот, олигомеры совмещаются легче. Даже один и тот же полимер, но разных М (М отличались более, чем в 10 раз) при определенных температурах расслаивались как в присутствии растворителя, так и без него.
Предел расслаивания пары полимеров в общем растворителе уменьшается с ухудшением качества растворителя и концентрационная область существования однофазных смесей полимеров в растворе уменьшается при переходе к «плохим» растворителям.
Температура.
 Влияние температуры на совместимость полимеров зависит от вида фазовой диаграммы – с ВКТС или НКТС (верхняя критическая точка смешения и нижняя критическая точка смешения)
Влияние температуры на совместимость полимеров зависит от вида фазовой диаграммы – с ВКТС или НКТС (верхняя критическая точка смешения и нижняя критическая точка смешения)

 Т Ж Т
Т Ж Т

 Ж
Ж
А В А В
Рис.25. Фазовые диаграммы с верхней (ВКТС) и нижней (НКТС) критическими точками смешения полимеров.
Напряжение сдвига.
Сдвиг вызывает дробление капель. А на границе капель существует межфазный переходный слой. Когда диаметр капель становится соизмеримым с толщиной межфазного слоя, то достигается оптическая прозрачность и наступает полное исчезновение признаков фазового разделения. Это справедливо и к смесям полимеров без растворителя (расплав).
3.2.1. Влияние растворителя на структуру пленок смеси полимеров, получаемых из раствора.
При испарении растворителя концентрация полимеров в растворе возрастает вплоть до 100%. При повышении концентрации полимера достигается предел растворимости и совместимая при определенной концентрации система становится несовместимой – наступает расслаивание. Если подъём температуры происходит медленно, концентрация и вязкость системы нарастают медленно и разделение растворов полимеров будет происходить медленно. В результате образуются либо крупные капли (пленка мутная), либо полное разделение полимеров в пленке на два слоя и тогда пленка прозрачная.


 А
А
 В
В
Возможен третий вариант – образуются не капли одного полимера в матрице другого, а сетка одного полимера в матрице другого полимера.
Для кристаллизующихся полимеров природа растворителя может определять ту или иную кристаллическую форму. Так, поливинилиденфторид ПВФ-2 из раствора в циклогексаноне кристаллизуется в a -форме (симметрия цепи 21, проекция мономерного звена на ось цепи 2,33Ǻ), а из раствора в диметилформамиде – в g - форме (симметрия цепи Z, проекция 2,57Ǻ). Пленки, приготовленные из смеси ПВФ-2 и Ф42 (сополимер тетрафторэтилена и винилиденфторида) в циклогексаноне демонстрируют несовместимость этих полимеров (две Тпл и Ткр), а из раствора в диметилформамиде – совместимость (одна Тпл и Ткр). К сведению, у Ф - 42 симметрия цепи Z, проекция мономерного звена на ось цепи – 2,54А.
Сравните различия в проекциях мономерного звена на ось цепи в этих системах
| Проекция звена на ось цепи, Ǻ | a-ПВФ-2 – Ф42 2,33 2,54 |  ПВФ-2 – Ф42 2,57 2,54 ПВФ-2 – Ф42 2,57 2,54 |
| .Ǻ | 0,21 | 0,03 |
| ,% | 8,26 | 1,18 |
Кстати, при растяжении пленок и волокон из смеси g-ПВФ-2/Ф-42 вероятно образуется сетка ПВФ – 2 в матрице Ф - 42, что позволило осуществить вытяжку выше Тпл Ф - 42 и существенно повысить прочность (>50%).
Если проводить не испарение растворителя, а его сублимацию, структура исходного раствора сохраняется в большей степени. Это касается размера частиц дисперсной фазы. А размер дисперсной фазы влияет на физико-механические свойства - 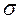 р уменьшается с увеличением размера частиц.
р уменьшается с увеличением размера частиц.
3.3. Структура твёрдых растворов полимеров.
В тех случаях, когда кристаллизующиеся полимеры образуют твёрдый раствор (изоморфизм замещения), образующиеся кристаллические структуры внешне не отличаются от тех, которые образуются в индивидуальных кристаллизующихся полимерах, т.е. обычно являются сферолитами. Первичным структурным элементом в неориентированных образцах являются ламели, образованные молекулярными цепями обоих компонентов. Величина складки индивидуальных полимеров различна, поэтому, естественно, в ламеле имеются области, построенные преимущественно макромолекулами каждого сорта. Следовательно, в отличие от твёрдых растворов низкомолекулярных веществ, в изоморфно-замещённых полимерных кристаллах наблюдается не статистическое распределение макромолекул разного сорта, а статистическое распределение областей макромолекул разного сорта. Модель строения твёрдого раствора полимеров представлена на рис. 26.
б
Рис.26. Модель строения твёрдого раствора. а– общий вид ламели; б – разрез ламели по плоскости, перпендикулярной длине складок.
Упорядоченные области (кристаллиты) внутри ламели разделены менее упорядоченными (аморфными) областями, распределение макромолекул обоих сортов в которых также не является статистическим. Наряду с участками, состоящими из беспорядочно чередующихся макромолекул, имеются участки, состоящие из макромолекул каждого сорта. Распределение проходных цепей, соединяющих различные ламели, также не является статистическим. Этим обусловлено обнаружение в таких системах трёх температур стеклования – смешиваемых полимеров и их твёрдого раствора.
Напряжения, возникающие при одноосной деформации, распределяются неравномерно между имеющимися в полимере проходными цепями. Нагруженные проходные цепи одного сорта как бы вытаскивают из изоморфнозамещённого кристаллита свои складки макромолекул, которые, ориентируясь вдоль силового поля, самостоятельно кристаллизуются. Этот процесс приводит первоначально однофазную систему к разделению на две кристаллические фазы. Это и позволило осуществить ориентационную термовытяжку волокон из смеси Ф-2/Ф-42 выше температуры плавления Ф-42.
3.4. Механизм смешения несовместимых полимеров.
Теории смешения рассматривают простой сдвиг двух несмешивающихся жидкостей, приводящий к деформации капли (рис. 27).
Растяжение
 | |||||||||||||||||||
 | |||||||||||||||||||
 | 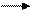 |  | |||||||||||||||||
 | |||||||||||||||||||
 | |||||||||||||||||||
 |  | ||||||||||||||||||
 | |||||||||||||||||||
 |
Сжатие
Характер деформирования зависит от вязкости капли h1 и матрицы h2: λ = ή1/ ή2 (рис.18).

 Скорость деформирования
Скорость деформирования
Рис. 27. Характер деформирования вязкой капли.
Разрушение также происходит при остановке потока. В момент остановки потока капля, стабильная при заданном градиенте скорости, распадается на множество мелких капель.
Все эти соображения относятся к ньютоновским жидкостям. Смешение полимеров в вязкотекучем состоянии отличается от смешения в растворе тем, что вязкость расплавов в тысячи раз превышает вязкость растворов. Как следствие этого, образование нитевидных структур происходит при значительно меньших скоростях сдвига и зависит от размера частиц: в первую очередь деформируются, вытягивается в нити более крупные частицы. По мере увеличения скорости сдвига в этот процесс вовлекаются мелкие частицы, а крупные, волокноподобные начинают разрушаться на мелкие фрагменты. Специфика разрушения полимерных вязкоупругих систем обусловлена способностью полимерной жидкости к ориентации и упрочнению, что приводит к возникновению полидисперсности образующихся капель.
3.5. Типы фазовых структур в смесях несовместимых полимера.
Высокая вязкость полимера приводит к тому, что равновесная форма частиц дисперсной фазы не возникает при сколь угодно длительном хранении или даже при специальном прогреве – «отжиге». Если же хотя бы один из компонентов смеси при охлаждении после смесительного аппарата кристаллизуется или стеклуется, то приближение к равновесию тем более затруднено. В результате образуется большое разнообразие форм фазовых структур (рис. 28).
дисперсия волокна коаксиальные микроэмульсия взаимо-
цилиндры в диспенрсной проникающие
фазе сетки (матрица
в матрице)
Рис.28.
P.S. Запатентован способ получения сверхтонких коротких волокон путем растворения матрицы в смеси со структурой 2 – волокна.
3.6. Коллоидно-химическая структура смесей несовместимых полимеров.
Большинство смесей полимеров двухфазны и их можно рассматривать, как коллоидные системы типа «полимер в полимере». Они обладают высокой агрегативной устойчивостью благодаря огромной вязкости, малому межфазному натяжению, развитому межфазному слою и пр.
Механические свойства двухфазной смеси полимеров зависят от:
1) соотношения объёмов дисперсной фазы и дисперсионной среды;
2) природы фаз;
3) размеров и формы частиц дисперсной фазы;
4) интенсивности взаимодействия полимеров на границе раздела фаз.
В смесях полимеров может наблюдаться снижение или увеличение Тс, ускорение или замедление кристаллизации, увеличение или уменьшение температуры плавления и пр. Конечный эффект присутствия второго полимера в смеси не однозначен. Так, при малых размерах частиц дисперсной фазы (~0,025 мкм) прочность возрастает, а при увеличении размеров частиц (~0,25 мкм) – уменьшается. Однако и чрезмерное дробление дисперсной фазы нежелательно.
Если смесь полимеров одноосно деформируют в вязкотекучем состоянии (экструзия), то частички дисперсной фазы изменяют форму – оптимальная форма - волокно. Это позволяет создавать напряженные конструкции (типа железобетона), в которых прочность на 15-20% выше. Например, ПВХ и СКН. Трубки из такой резины обладают термоусадкой. Их используют для изоляции проводов, спаев проводов и т.п.
Как уже указывалось ранее, в основном физико-механические свойства композиции зависят от природы непрерывной фазы. При изменении соотношения полимеров может происходить так называемое «обращение фаз». Обычно этот переход происходит при увеличении количества добавляемого компонента выше 50%. Однако уровень вязкости смешиваемых полимеров и способ приготовления смеси может вызвать неполное разделение фаз с образованием двух непрерывных фаз (сетка в сетке). В этом случае две непрерывные фазы могут наблюдаться в интервале 30-70% содержания каждого полимера. Если же вязкость одного из полимеров значительно меньше вязкости другого, то маловязкий полимер может образовывать коагуляционный или конденсационный каркас уже при содержании ³ 10% (коагуляционный – слияние частиц, разделённых прослойками диспесионной среды; конденсационный - непосредственная связь между дисперсными частицами.)

Рис.29. Типы контактов между твёрдыми фазами
Если температурная зависимость вязкости у смешиваемых полимеров существенно различна, может создаться ситуация, когда, в зависимости от температуры, менее вязким полимером станет тот или иной. Например, можно создать пару полимеров ПММА и ПИБ так, чтобы ПММА при 1600С был более вязким, а при 1800С – менее вязким, чем ПИБ. Это приводит к тому, что при температуре смешения 180оС непрерывной фазой оказывается ПММА, а при 1600С – ПИБ. Доказательством этого служит селективное растворение, например, ПММА в метилэтилкетоне. Смесь, приготовленная при 1800С, после растворения ПММА представляет собой порошок, а приготовленная при 1600С сохраняет целостность. Этот результат имеет технологическое значение.
3.6.1. Межфазный слой.
Граница раздела полимерных фаз – это не поверхность, а слой определенной толщины  , зависящий от свойств контактирующих фаз. Увеличение поверхностного натяжения (уменьшение термодинамического сродства фаз) приводит к уменьшению толщины поверхностного слоя, и наоборот.
, зависящий от свойств контактирующих фаз. Увеличение поверхностного натяжения (уменьшение термодинамического сродства фаз) приводит к уменьшению толщины поверхностного слоя, и наоборот.
Межфазный слой на границе раздела полимер – полимер имеет особенности строения, обусловленные природой контактирующих фаз. Как известно, мономеры смешиваются, растворяются друг в друге неограниченно, олигомеры сорастворимы при М ~ Мсегмента, полимеры в подавляющем числе случаев несовместимы, не растворяются друг в друге. Однако, на границе раздела полимерных фаз происходит сорастворимость сегментов и межфазный слой, таким образом, представляет собой как бы раствор двух олигомеров.
Установлено, что состав межфазного слоя меняется монотонно, ассимптотически приближаясь к составу соответствующей фазы. Аналогично изменяется плотность слоя, при этом в слое плотность ниже аддитивной.
3.7. Особенности физико-механических свойств смесей полимеров.
Стеклование.
Если смесь полимеров двухфазна, то проявляются две Тс, соответствующих исходным полимерам. Если смесь однофазна, то наблюдается либо одна Тс, либо три – исходных полимеров и однофазной смеси. Положение Тс однофазной смеси на температурной шкале зависит от состава смеси.
Плавление и кристаллизация.
В твёрдых растворах полимеров в принципе должна была бы быть одна промежуточная Ткр и Тпл. Однако, как правило, обнаруживаются и температуры фазовых переходов исходных полимеров.
В двухфазный смесях кристаллизующихся полимеров может происходить замедление кристаллизации компонентов.
Плотность.
Плотность полимеров не зависит от их совместимости, а главным образом – от способа приготовления смеси и может быть как выше, так и ниже аддитивной.
Диффузия газов и жидкостей.
Введение полимера с пониженной газопроницаемостью уменьшает газопроницаемость основного полимера. Таким образом можно регулировать показатели газопроницаемости.
Прозрачность смеси аморфных полимеров определяется размером частиц дисперсной фазы и соотношением показателей преломления смешиваемых компонентов. Когда размер частиц становится сопоставимым с длиной волны l, прозрачность смеси увеличивается.
В области размеров частиц, обеспечивающих дифракцию световых лучей, последние рассеиваются под разными углами в зависимости от l. Это, как для любой коллоидной системы, приводит к опалесценции дисперсной системы. Опалесценция выражается в том, что система кажется более голубой в отраженном и более оранжевой в проходящем белом свете.
Горючесть.
Термодеструкция и воспламеняемость смеси снижается при добавлении термостабильного или трудновоспламеняющегося (негорючего) полимера. Эффект максимален тогда, когда «негорючий» полимер образует непрерывную фазу в смеси.
Деформация.
В двухфазный смесях полимеров, особенно, если модули упругости компонентов сильно отличаются, при деформации может происходить отслаивание матрицы от частиц дисперсной фазы. Если же модули упругости близки, отслаивания не происходит вплоть до больших деформаций.
Однофазные смеси аморфных полимеров деформируются как однородные системы.
В однофазных смесях вязкость от состава описываются уравнением Аррениуса:
lnh = φ1lnh1 + φ2 lnh2
Здесь φ1 и φ2 – мольные доли компонентов (φ1 + φ2 = 1), h1 и h2 - вязкость жидких компонентов смеси.
Формула Аррениуса означает логарифмическую адитивность вязкости, когда состав смеси выражен в мольных долях.
Существуют и другие формулы.
В целом, реологические свойства смеси полимеров определяется в основном свойствами полимера, образующего непрерывную фазу.
Релаксация напряжений и ползучесть также определяются главным образом свойствами полимера, образующего матрицу.
Прочность и разрушение.
Вообще, всякая неоднородность, в том числе и дисперсная фаза в смеси полимеров, является концентратором напряжения и снижает прочность.
Увеличение прочности может быть вызвано следующими причинами:
1. изменением направления роста трещины при встречи с частицей наполнителя;
2. ориентацией и упрочнением полимера в межфазном слое на границе раздела полимер – наполнитель;
3. возникновение собственной структуры наполнителя;
4. релаксация напряжения в вершине трещины в межфазном слое;
5. причины увеличения прочности – рассмотренные ранее (взаимопроникающие сетки – увеличение ориентации при вытяжке выше Тпл основного полимера и т.д.)

