 2015-01-30
2015-01-30 38662
38662Газовая хроматография представляет собой процесс разделения компонентов смеси, основанный на различии в равновесном распределении компонентов между двумя фазами – газом-носителем (подвижная фаза) и либо твердой фазой, либо жидкостью, нанесенной в виде тонкой пленки на поверхность твердого носителя или стенки хроматографической колонки (неподвижная фаза).
В первом случае метод называется газоадсорбционной хроматографией, во втором – газо-жидкостной распределительной хроматографией. Из этих двух вариантов газовой хроматографии наиболее распространена распределительная газожидкостная хроматография, которая и рассматривается далее.
Общие сведения о газожидкостной распределительной хроматографии
Сущность метода газожидкостной хроматографии (ГЖХ) состоит в следующем. Анализируемая смесь летучих компонентов (обычно – раствор) переводится в парообразное состояние и смешивается с потоком инертного газаносителя, образуя с ним подвижную фазу. Эта смесь проталкивается далее новой порцией непрерывно подаваемого газа-носителя и попадается в хроматографическую колонку, заполненную неподвижной (стационарной) жидкой фазой.
Разделяемые компоненты распределяются между фазами в соответствии с их коэффициентами распределения Kр, определяемыми по уравнению (3.5). Равновесный обмен хроматографируемого вещества между подвижной и неподвижной фазами осуществляется в результате многократного повторения актов сорбция ↔ десорбция по мере движения подвижной фазы вдоль неподвижной внутри хроматографической колонки.
Поток газаносителя увлекает с собой разделяемую парообразную смесь вдоль хроматографической колонки, так что процессы сорбция ↔ десорбция разделяемых компонентов повторяются многократно, причем каждый раз в системе между фазами устанавливается динамическое равновесие. Эти многократные переходы разделяемых веществ из подвижной фазы в неподвижную и обратно совершаются по всей длине хроматографической колонки до тех пор, по пары разделяемых веществ не покинут колонку вместе с газомносителем.
Поскольку сродство различных разделяемых веществ к неподвижной фазе различно, то в процессе сорбционно-десорбционных процессов они задерживаются в неподвижной фазе неодинаковое время. Чем выше температура кипения и относительная растворимость вещества в неподвижной фазе, тем дольше оно в ней находится, тем позже покидает хроматографическую колонку. В конце концов из хроматографической колонки вместе с газомносителем выходят зоны (объемы) парообразных хроматографируемых веществ, разделенных полностью или частично.
Если для двух компонентов смеси коэффициенты распределения Kр одинаковы, то они не разделяются. Если же их коэффициенты распределения различны, то разделение происходит, причем первым покидает колонку тот компонент, у которого температура кипения ниже.
Пары разделенных компонентов вместе с газомносителем поступают в детектор хроматографа, генерирующий электрический сигнал – тем больший, чем выше концентрация компонента в парогазовой смеси. Электрический сигнал усиливается и фиксируется регистратором хроматографа в виде хроматограммы, записываемой на диаграммной ленте. Эти хроматограммы и используются для качественной и количественной обработки результатов анализа разделяемой смеси компонентов.
Основные характеристики хроматограммы
Параметры удерживания
Хроматограмма – это зарегистрированная во времени последовательность показаний регистратора. Каждому разделенному компоненту смеси соответствует свой пик на хроматограмме. По оси абсцисс откладывается время, по оси ординат – величина аналитического сигнала, которая тем больше, чем выше содержание данного компонента в разделяемой смеси.
На рис. 3.6 схематично показан общий вид хроматограммы в случае разделения трехкомпонентной смеси, состоящей из компонентов А и В, сорбируемых в колонке, и компонента, не сорбируемого в колонке. Каждому из трех компонентов на хроматограмме отвечает свой пик. Значение t = 0 соответствует моменту ввода пробы, от которого начинает отсчитываться время t.
Величина tA – время удерживания компонента А, tВ – время удерживания компонента В, t0 – время выхода несорбируемого компонента. В данном случае оба компонента А и В разделяются полностью, поэтому их пики на хроматограмме не накладываются друг на друга.
Время удерживания – качественная характеристика каждого компонента. Оно измеряется от момента ввода пробы до момента выхода максимума (вершины) пика. Время удерживания зависит от природы хроматографируемого вещества и газаносителя, скорости прохождения подвижной фазы через хроматографическую колонку, от природы и массы неподвижной фазы, температуры, длины колонки.
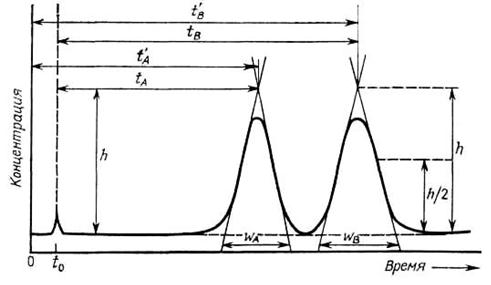
Рис. 3.6. Основные параметры хроматограммы
Время выхода t0 несорбируемого компонента (например, растворителя) определяется соотношением
t0 = L / U,
где L – длина хроматографической колонки; U – линейная скорость движения потока газа-носителя.
Исправленное время удерживания компонентов А и В (соответственно tA и tB), равное
tA = tA – t0, tВ = tВ – t0,
– это время, в течение которого данный компонент находился в неподвижной фазе. Исправленное время удерживания пропорционально коэффициенту распределения Kр данного компонента разделяемой смеси.
Относительное время удерживания tr и относительное исправленное время удерживания tr определяются формулами
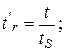
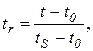 (3.10)
(3.10)
где t – время удерживания данного вещества; tS – время удерживания стандартного вещества (стандарта); t0 – время выхода несорбируемого компонента при хроматографировании веществ в одних и тех же условиях. Относительное время удерживания меньше зависит от внешних условий, чем время удерживания.
На рис. 3.6 также показаны ширина пиков у их основания (wA и wB), высота (h) и полувысота (h /2) пиков. При анализе хроматограмм иногда определяют ширину пиков на середине их высоты – wA, ½ и wB, ½.
На практике часто измеряют не время удерживания, а расстояние удерживания l, пропорциональное времени удерживания, т.е. расстояние (например, в мм) на хроматограмме от точки, соответствующей моменту ввода пробы, до абсциссы, отвечающей положению максимума (вершины) пика.
Кроме времени удерживания иногда используют такой параметр, как объем удерживания (удерживаемый объем), равный объему подвижной фазы, который выносит из колонки все данное вещество. Объем удерживания V зависит от скорости U движения подвижной фазы и равен произведению времени удерживания t на эту скорость:
V = t · U.
Коэффициент удерживания (замедления) R – это отношение скорости перемещения w данного компонента вдоль хроматографической колонки к скорости U движения потока газа-носителя:
R = w / U.
Поскольку
w = L / t и U = L / t0,
где L – длина колонки; t – время удерживания данного компонента; t0 – время выхода несорбируемого компонента, то
R = t0 / t.
Коэффициент емкости k равен отношению исправленного времени удерживания t = t – t0 данного компонента к t0:
R = (t – t0) / t0 .
Чем выше k, тем большее время находится данный компонент в неподвижной фазе.
Параметры разделения. Эффективность колонки
К параметрам разделения двух веществ относятся степень и коэффициент разделения. Эффективность хроматографической колонки характеризуется числом теоретических тарелок и величиной, эквивалентной теоретической тарелке.
Степень разделения RS (разрешение пиков) количественно характеризует разделение двух пиков на хроматограмме и рассчитывается по формуле, аналогичной формуле (3.8) для плоскостной хроматограммы:
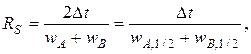
где t = tB – tA – разность времен удерживания разделяемых компонентов А и В; wA и wB – ширина пиков; wA, ½ и wB, ½ – полуширина пиков.
Если RS < 1, то разделение двух веществ неполное. При RS > 1 наблюдается полное разделение двух компонентов смеси (рис. 3.7).
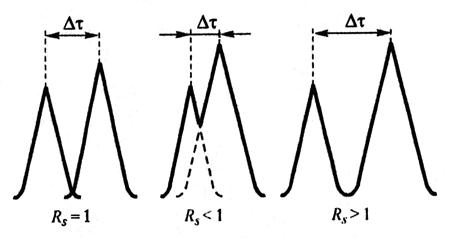
Рис. 3.7. Разделение t пиков на хроматограмме при различных значениях RS
Разделение пиков t прямо пропорционально длине L хроматографической колонки, тогда как сумма полуширин пиков прямо пропорциональна корню квадратному из L:
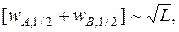
поэтому степень разделения RS также оказывается прямо пропорциональной длине колонки. Отсюда следует, что с ростом длины колонки L степень разделения RS увеличивается, однако одновременно возрастает и продолжительность анализа.
Степень разделения в ГЖХ зависит от таких хроматографических параметров, как коэффициент разделения α и число теоретических тарелок n.
Коэффициент разделения α рассчитывается по формуле:
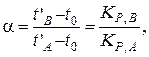
где tA и tB – соответственно время удерживания компонентов А и В; t0 – время выхода несорбируемого компонента; КР,А и КР,В – коэффициенты распределения компонентов А и В соответственно.
Коэффициент разделения характеризует селективность неподвижной фазы по отношению к двум данным компонентам и относительное расположение разделяемых пиков на хроматограмме. Коэффициент разделения α и степень разделения RS связаны соотношением
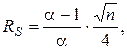
где n – так называемое число теоретических тарелок.
Если α = 1, то RS = 0, т.е. два хроматографируемых вещества не разделяются. Чем больше величина α, тем лучше разделение пиков на хроматограмме, тем неподвижная фаза более селективна по отношению к двум данным разделяемым веществам.
Число теоретических тарелок n. При хроматографическом разделении компонентов смеси осуществляется перенос вещества через границу раздела двух фаз – подвижной и неподвижной. Чем больше число таких переходов, тем более полно разделяются компоненты смеси. Количество подобных переходов характеризует эффективность хроматографической колонки.
Участок зоны внутри колонки, на котором устанавливается равновесное распределение данного вещества между подвижной и неподвижной фазами (сорбция ↔ десорбция) называют теоретической тарелкой (по аналогии с терминологией, принятой в теории ректификации для ректификационных колонн, в которых осуществляются многократно повторяемые акты испарение ↔ конденсация).
Число теоретических тарелок n рассчитывается по формуле
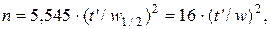
где t – время (или расстояние) удерживания данного компонента смеси; w и w 1/2 – соответственно ширина и полуширина пика, выраженная в тех же единицах, что и t.
Чем больше теоретических тарелок n, тем эффективнее работа хроматографической колонки. Число теоретических тарелок может быть от нескольких сотен до нескольких тысяч.
Если длина хроматографической колонки составляет L, а число теоретических тарелок равно n, то высота, эквивалентная теоретической тарелке, (ВЭТТ) H рассчитывается по формуле
H = L / n.
Чем меньше ВЭТТ, тем менее размыта зона (полоса) отделяемого компонента при его выходе из колонки. Величина ВЭТТ в оптимальном случае часто не превышает 1,5 мм, хотя может быть и несколько большей.
Параметры Н и n характеризуют эффективность хроматографической колонки при разделении смеси компонентов. Чем больше n и меньше Н, тем полнее отделение зоны (полосы) данного компонента от зон остальных компонентов при их разделении.
Разработаны теоретические подходы, позволяющие повысить эффективность ГЖХ-разделения – уменьшить степень размывания зоны разделяемого компонента. При этом учитывается роль вихревой и молекулярной диффузии, сопротивление системы массопереносу веществ и другие факторы. Ван-Деемтер предложил уравнение, позволяющее на основе разработанных подходов рассчитывать величину ВЭТТ (уравнение Ван-Деемтера):
Н = А + В / U + СU, (3.11)
где А, В, С – коэффициенты, учитывающие вклад соответственно вихревой диффузии, молекулярной диффузии и сопротивления массопереносу в размывание зоны хроматографируемого компонента; U – линейная скорость потока газаносителя.
Константа А связана с действием вихревой диффузии, которая зависит от размера частиц и плотности заполнения колонки. Величина В связана с коэффициентом диффузии молекул в подвижной фазе. Слагаемое В / U учитывает действие продольной диффузии. Постоянная С характеризует кинетику процесса сорбция ↔ десорбция, массопередачу и другие эффекты. Влияние каждого слагаемого уравнения (3.11) на величину Н в зависимости от скорости подвижной фазы показано на рис. 3.8.
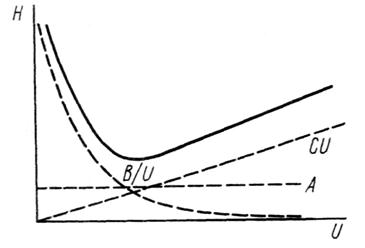
Рис. 3.8. Зависимость высоты, эквивалентной теоретической тарелке, Н от скорости подвижной фазы U.
Первое слагаемое дает постоянный вклад в Н. Вклад второго слагаемого заметен при небольшой скорости потока. С увеличением скорости подвижной фазы влияние третьего слагаемого возрастает, а доля второго уменьшается. Суммарная кривая, характеризующая зависимость Н от скорости потока, представляет собой гиперболу.
При небольшой скорости потока высота, эквивалентная теоретической тарелке, уменьшается, а затем начинает возрастать. Поскольку эффективность колонки тем выше, чем меньше ВЭТТ, оптимальная скорость подвижной фазы будет равна скорости, соответствующей точке минимума на этой кривой. Чтобы найти эту точку, продифференцируем уравнение (3.11) и производную приравняем к нулю:
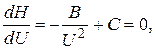
откуда
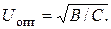
Подставляя выражение для U опт в (3.11), находим оптимальную высоту, эквивалентную теоретической тарелке:
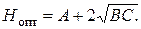
Таким образом, динамическая теория дает основу для оптимизации хроматографического процесса.
Основные узлы и используемые материалы газожидкостного хроматографа
Отечественная промышленность и зарубежные фирмы выпускают большое количество хроматографов самых различных типов. Независимо от сложности устройства основными узлами хроматографической установки являются дозатор (система ввода пробы), хроматографическая колонка и детектор. Кроме того, в установке имеются устройства для подачи газаносителя, для преобразования импульса детектора в соответствующий сигнал и некоторые другие.
Газыносители
В качестве газовносителей используются инертные газы (гелий, аргон), а также азот, диоксид углерода и водород. Выбор газаносителя отчасти определяется детектором. Для удаления следов воды газ иногда пропускают через молекулярные сита. Поток газа обеспечивается избыточным давлением газового баллона, поэтому можно работать без насоса. Чтобы получать воспроизводимые результаты измерений, поток носителя следует поддерживать неизменным.
При работе в изотермическом режиме достаточно установить давление на колонке с помощью двухступенчатого кранаредуктора. При программировании температуры необходимо использовать регулятор потока. Для измерения скорости потока на входе в колонку может быть использован ротаметр, а на выходе – мыльно-пузырьковый измеритель потока. Для набивных колонок расход варьируют между 25 и 150 см3/мин, а для капиллярных – в пределах 1 – 25 см3/мин.
Система ввода пробы
Газообразные и жидкие пробы обычно вводят с помощью специальных шприцев, прокалывая в месте ввода пробы диафрагму из силиконовой резины (септу). Для газообразных проб применяются газовые шприцы, для жидких – микрошприцы. Микрошприцы позволяют вводить в хроматограф пробы объемом от долей до десятков микролитров. Твердые пробы вводят в хроматограф или после перевода их в раствор, или непосредственным испарением пробы в нагреваемом дозаторе, куда она вводится с помощью игольного ушка. Температура испарителя обычно задается примерно на 50 оС выше температуры кипения наименее летучего компонента вводимой смеси.
Хроматографические колонки
Хроматографические колонки весьма различны по форме, размерам и конструкционным материалам. Применяются прямые, спиральные и другие колонки длиной от одногодвух до нескольких десятков метров. Внутренний диаметр колонок составляет обычно несколько миллиметров. В зависимости от свойств анализируемой системы в качестве конструкционных материалов для колонок чаще всего используют сталь, латунь, медь, стекло и др. Материал колонки должен обладать определенной химической инертностью по отношению к компонентам пробы.
Адсорбент, наполняющий колонку, должен обладать рядом свойств: необходимой селективностью, достаточной механической прочностью, химической инертностью к компонентам смеси и быть доступным. На практике в качестве адсорбентов обычно используют оксид алюминия, силикагели, активированные угли, пористые сополимеры на основе стирола и дивинилбензола, синтетические цеолиты. Широко используют модифицированные адсорбенты, которые получают обработкой исходных адсорбентов растворами кислот, щелочей, неорганических солей и т.д. Выбор адсорбента зависит от методики хроматографирования.
Жидкости, используемые в качестве неподвижных фаз в ГЖХ, должны быть термически и химически устойчивыми, малолетучими, чтобы не происходила их десорбция из колонки. Температура кипения неподвижной фазы – примерно на 100 оС выше требуемой температуры колонки. Разделяющая жидкость должна обладать определенной селективностью, т.е. обеспечивать различные коэффициенты распределения для определяемых веществ. Однако эти коэффициенты не должны быть ни слишком большими, ни слишком малыми, иначе удерживание соединений будет слишком сильным или настолько слабым, что не произойдет разделения.
Предложены многие десятки и даже сотни вариантов жидких неподвижных фаз, отвечающих перечисленным выше требованиям. Это углеводороды (индивидуальные или смеси) с числом углеродных атомов в цепи от 10 до 30, полисилоксаны (силиконы), полиэтиленгликоли, полиэфиры, амиды, амины, жирные кислоты и др. Масса жидкой неподвижной фазы обычно составляет от 1 до 20 % от массы твердого носителя.
Поскольку большое влияние на процесс хроматографического разделения оказывает температура, хроматографические колонки, как правило, термостатируются. Для этого используется их обогрев жидкостью (или парами кипящей жидкости), воздушное термостатирование или какой-либо другой прием.
Детекторы
Детектор предназначен для обнаружения изменений в составе газа, прошедшего через колонку. Показания детектора обычно преобразуются в электрический сигнал и передаются фиксирующему или записывающему прибору. Основными характеристиками детектора являются чувствительность, пределы детектирования, инерционность и диапазон линейной зависимости между концентрацией и величиной сигнала.
Существующие способы детектирования и сами детекторы подразделяют на дифференциальные и интегральные. Дифференциальные передают мгновенное значение некоторой характеристики, интегральные – суммируют изменение этой характеристики за определенный промежуток времени.
К группе дифференциальных относятся детекторы по теплопроводности (катарометр), пламенноионизационный (ПИД), термоионный (ТИД), пламеннофотометрический (ПФД), электронозахватный (ЭЗД) и др. Общая характеристика детекторов для газожидкостной хроматографии приведена в табл. 3.5.
Таблица 3.5
Дифференциальные детекторы для газожидкостной хроматографии
| Детектор | Детектируемые компоненты | Предел обнаружения | Диапазон линейности |
| Катарометр | Неизбирателен | 10–8 г/см3 | 104 |
| ПИД | Соединения, содержащие углерод и водород | 10–13 г/с | 107 |
| ТИД | Р N | 1015 г/с 1014 г/с | 105 |
| ПФД | P S | 3·10–13 г/с 2·10–11 г/с | 105 |
| ЭЗД | Электроотрицательные группы | 5·10–14 г/с | 5·104 |
Дифференциальные детекторы подразделяют на концентрационные и потоковые. Концентрационные регистрируют концентрацию, потоковые – произведение концентрации на скорость, т.е. поток вещества. К концентрационным относятся детекторы, показания которых зависят от скорости потока (катарометр, газовые весы, детектор по ионизации βизлучением и др.). К потоковым относятся детекторы, показания которых не зависят от скорости потока (термохимический детектор, пламенноионизационный и т.д.).
Сигналы интегральных детекторов, работа которых основана на титровании или поглощении газаносителя, пропорциональны общей массе вещества в элюированной полосе, поэтому их называют массовыми детекторами.
Если показания детекторов линейны, то имеют место следующие зависимости значения сигнала.
1. Для концентрационного детектора сигнал Ес пропорционален концентрации:
Ес = φ с · с,
где φ с – коэффициент пропорциональности, характеризующий чувствительность концентрационного детектора; с – концентрация.
Так как с = d m /d V, а объемная скорость газа α S = d V /d t (V – объем газа; t – время; m – масса вещества), то
d m = c · d V = c · α S · d t,
или
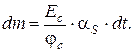
Количество анализируемого компонента за время от t 1 до t 2 при постоянной скорости определяется соотношением:

Чтобы определить m, надо найти площадь пика на хроматограмме: Ec = h · C 1 (C 1 – чувствительность самописца, мВ/см); t = l / u (u – скорость диаграммной ленты; l – расстояние на диаграммной ленте, соответствующее времени t; h – отклонение пера самописца). Тогда
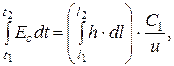 (3.12)
(3.12)
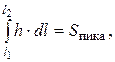 (3.13)
(3.13)
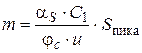 .
.
Для концентрационного детектора необходимо, чтобы расход был постоянным. Если это не соблюдается, то нет пропорциональности между количеством вещества и площадью пика, а значит определение концентрации по площади пика неверно. Поэтому при работе с концентрационными детекторами аналитическим сигналом служит высота пика, которая не зависит от скорости потока.
2. Для потокового детектора сигнал определяется числом молекул, достигших чувствительного элемента в данный момент. Обозначим через j поток анализируемого компонента, т.е. количество вещества, проходящего в единицу времени через единицу сечения:
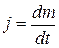 .
.
Тогда
Еj = φ j · j,
или
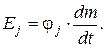
Отсюда
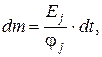
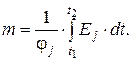
С учетом (3.12) и (3.13)
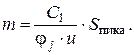
Для потокового детектора сигнал пропорционален количеству вещества и не зависит от расхода газаносителя (площадь пика остается постоянной, а высота увеличивается с повышением скорости). Таким образом, при работе с потоковыми детекторами аналитическим сигналом служит площадь пика.
3. Для интегральных детекторов, у которых сигнал определяется полным числом молекул, попадающих к чувствительным элементам,
Еm = φ m · m,
где Em – сигнал массового детектора; φ m – коэффициент пропорциональности, характеризующий чувствительность интегрального детектора; m – количество вещества, равное
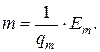
Существует связь между показаниями интегрального и дифференциального детекторов:
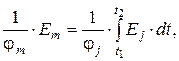
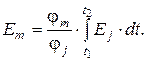
Итак, для потокового детектора с уменьшением скорости потока высота пиков падает, ширина растет, площадь постоянна. Для концентрационного детектора высота пиков постоянна, а площадь растет за счет роста ширины.
По зависимости площади пика на хроматограмме от скорости потока можно определить тип детектора.
Общая формула зависимости сигнала от чувствительности, концентрации и расхода:
E = φ · c · α S n.
Если n = 1, то детектор потоковый (j = c · α S, и E = φ j · j).
Если n = 0, то детектор концентрационный (E = φ с · c).
Если n ≠ 1, то детектор промежуточный.
Лучший детектор – потоковый, так как его показания не зависят от расхода газаносителя и слабо зависят от температуры.
В интегральных детекторах анализируемый газ на выходе из колонки поглощается каким-либо раствором, а затем анализируется или поглощающий раствор или оставшийся непоглощенный газ. Если носителем является диоксид углерода, то после колонки газ барботируется через раствор щелочи и измеряется объем газа, не поглощенного этой жидкостью.
Достоинствами интегральных детекторов является их простота и широкая область линейной зависимости показаний детектора от количества вещества. К недостаткам относятся значительная инерционность и низкая чувствительность, в связи с чем такие детекторы в настоящее время применяются редко.
Одним из наиболее распространенных дифференциальных детекторов является катарометр. Он представляет собой массивный блок из латуни или нержавеющей стали (рис. 3.9) с двумя ячейками, в каждой из которых находятся чувствительные нагревательные элементы – нити из вольфрамовой или платиновой проволоки.
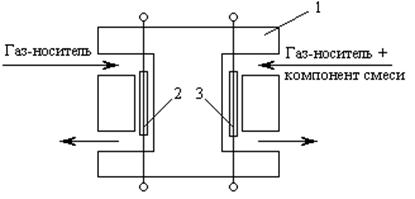
Р ис. 3.9. Схема ячейки детектора по теплопроводности:
(катарометра): 1 – корпус; 2, 3 – нагревательные элементы
Принцип работы катарометра заключается в следующем. Нагревательные элементы в сравнительной и рабочей ячейках нагревают постоянным электрическим током. Теплопроводность окружающего их газа определяет температуру, следовательно, и сопротивление нагревательных элементов. Когда через обе ячейки катарометра протекает чистый газноситель, температура нагревательных элементов одинакова.
Если через сравнительную ячейку катарометра протекает чистый газноситель, а через измерительную – газноситель плюс компонент, выходящий из хроматографической колонки, то температура, следовательно, и сопротивление нагревательных элементов будут разные, что нарушает баланс измерительного моста (рис. 3.10). Различие в температуре обусловлено различием в теплопроводности газа в сравнительной и измерительной ячейках катарометра.
В катарометре реализована мостовая схема Уитстона (рис. 3.10). Она содержит два вмонтированных нагревательных элемента R 1 и R 2 и два одинаковых проволочных сопротивления R 3 и R 4. Таким образом, чувствительные нагревательные элементы являются активными плечами мостовой измерительной схемы. На мост подается постоянное стабилизированное напряжение 6 – 12 В. Вследствие этого температура чувствительных элементов повышается до тех пор, пока не установится равновесие между подводимой электрической энергией и потерей теплоты.
Если мост в начале работы при продувании через обе ячейки газаносителя сбалансирован сопротивлением R 5, а затем к газуносителю, выходящему из хроматографической колонки, подмешивается какойлибо компонент, имеющий другую теплопроводность, то в мостовой схеме возникает разность потенциалов между клеммами А и В, обусловленная различием сопротивлений нагревательных элементов в сравнительной и измерительной ячейках. Возникшая разность потенциалов усиливается и фиксируется на ленте самописца регистратора в виде хроматограммы.
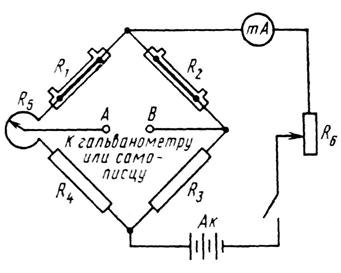
Рис. 3.10. Мостовая измерительная схема катарометра:
R 1, R 2 – нагревательные элементы; R 3, R 4 – проволочные стандартные сопротивления; R 5 – нулевой потенциометр; R 6 – токовый реостат; Ак – аккумуляторная батарея; mA – миллиамперметр
Количество теплоты, отводимое от нагретой нити при постоянных условиях, зависит от состава газа. Чем больше теплопроводность определяемых компонентов смеси будет отличаться от теплопроводности газаносителя, тем большей чувствительностью будет обладать катарометр. Наиболее подходящим газомносителем с этой точки зрения является водород, теплопроводность которого значительно превышает теплопроводность большинства других газов. В целях техники безопасности чаще применяется гелий, теплопроводность которого также достаточно высока.
В последнее время металлические нити в катарометре успешно заменяются термисторами, имеющими более высокий, чем у металлов, температурный коэффициент электрической проводимости. Достоинствами катарометра являются простота, достаточная точность и надежность в работе. Однако изза сравнительно невысокой чувствительности он не применяется для определения микропримесей.
Пламенноионизационные детекторы более чувствительны, чем детекторы по теплопроводности. Принцип их действия состоит в следующем. После разделения компонентов смеси подвижная фаза поступает из хроматографической колонки в пламя водородной лампы, находящееся между электродами. Органические вещества подвижной фазы сгорают в пламени с образованием ионизированных продуктов, вследствие чего возрастает электрический ток между электродами.
Увеличение электрической проводимости усиливается и фиксируется в виде записи хроматограммы на самописце регистратора. Высокая чувствительность детекторов этого типа обусловила их широкое применение. Однако высокая чувствительность ПИД проявляется только по отношению к органическим веществам. Чувствительность детектора по отношению к неорганическим соединениям (аммиак, сероводород, оксиды серы, кислород, азот и т.п.) резко падает.
Среди селективных детекторов наиболее широкое распространение получили пламеннофотометрический, термоионный и электроннозахватный.
Принцип действия пламеннофотометрического детектора основан на измерении интенсивности излучения продуктов атомизации компонентов подвижной фазы в водородном пламени. ПФД позволяет с высокой чувствительностью определять серосодержащие и фосфорорганические соединения (светопоглощение – соответственно при 394+ 10 нм и 52+ 10 нм). В термоионном детекторе в пламя горелки вводят соли щелочных металлов. При попадании в такое пламя соединений фосфора появляется ионный ток, пропорциональный содержанию атомов фосфора.
ТИД – селективный фосфорный детектор высокой чувствительности. Принцип работы электроннозахватного детектора (ЭЗД) близок к принципу действия пропорционального счетчика для измерения рентгеновского излучения. Под действием βизлучателей, таких, как 63Ni или тритий, в потоке газаносителя происходит ионизация и появляются электроны. При отсутствии детектируемых соединений ток, протекающий через ячейку, остается постоянным. В присутствии органических соединений, особенно, если они могут захватывать электроны, уровень тока уменьшается. ЭЗД является высокочувствительным специальным детектором на вещества, содержащие электроотрицательные группы – галогены, пероксиды, хиноны, фталаты, нитрогруппы и т.п.
Качественный анализ методом газо-жидкостной хроматографии
Качественный анализ, т.е. идентификация разделяемых компонентов с помощью хроматографической методики проводится преимущественно двумя методами: с использованием веществсвидетелей и времени удерживания.
Метод использования веществ-свидетелей
В тех же условиях, в которых получают хроматограмму разделяемой смеси, записывают хроматограммы веществсвидетелей, наличие которых предполагается в анализируемой смеси. Фиксируют время удерживания веществсвидетелей и сравнивают их со временем удерживания компонентов разделяемой смеси. Совпадение на хроматограмме разделяемой смеси времени удерживания веществасвидетеля со временем удерживания того или иного компонента может свидетельствовать о том, что данный компонент смеси и веществосвидетель идентичны.
Иногда веществосвидетель вносят непосредственно в пробу анализируемой смеси (метод метки). Записывают в одинаковых условиях хроматограммы такой пробы и пробы анализируемой смеси, не содержащей веществасвидетеля. Если число пиков остается одним и тем же, а интенсивность (высота) пика того или иного компонента на хроматограмме при внесении веществасвидетеля в пробу возрастает, то это означает, что данный компонент и веществосвидетель идентичны.
Некоторые родственные вещества могут иметь практически одинаковые времена удерживания при использовании данной хроматографической колонки с определенной неподвижной фазой. Для более надежной идентификации определяемых веществ следует проводить хроматографирование с использованием двух или нескольких неподвижных фаз различной полярности.
Метод относительных удерживаний
К анализируемой пробе прибавляют вещество сравнения и хроматографируют смесь строго в тех условиях, которые указаны в методике анализа. По формуле (3.10) определяют относительное исправленное время удерживания:
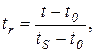
где t, tS, t0 – время удерживания соответственно определяемого компонента, вещества сравнения и несорбируемого компонента смеси. Сравнивают относительное исправленное время удерживания с указанным в методике.
Метод с использованием индексов удерживания Ковача
Обычно в таких условиях, когда описанные выше методы применить невозможно, для идентификации компонентов разделяемой смеси используют так называемые индексы удерживания Ковача – расчетные величины, определяемые на основании сравнения параметров удерживания близких по составу и строению веществ в предположении аддитивности изменения свойства в данном ряду родственных соединений.
Индексы удерживания Ковача определяют по формуле:
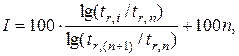
где tr – приведенное время удерживания; n – число атомов углерода в алкане; i – определяемое вещество.
Стандартом при определении индекса удерживания являются два соседних нормальных алкана, один из которых элюируется до, а второй после исследуемого соединения, т.е. tr,n < tr,i < tr,(n+ 1 ) .
Идентификация вещества по индексу удерживания производится путем хроматографирования соединения с последующим хроматографированием в тех же условиях двух соседних алканов, выбранных в качестве стандарта. Результаты анализа по индексу удерживания оказываются более надежными, чем по времени удерживания, так как индекс удерживания является более индивидуальной характеристикой вещества.
Индексы удерживания многих веществ при определенных температурах приводятся в соответствующих справочных таблицах, что облегчает проведение качественного анализа. Кроме того, накопленный экспериментальный материал позволил установить определенные зависимости между индексом удерживания и физикохимическими свойствами веществ.
Количественный анализ в хроматографии
Количественный хроматографический анализ основан на измерении различных параметров пика, зависящих от концентрации хроматографируемых веществ: высоты, ширины, площади, удерживаемого объема или произведения удерживаемого объема на высоту пика. При достаточной стабильности условий хроматографирования и детектирования определяющим параметром пика можно считать его высоту.
Расчет по площади пика позволяет несколько снизить требования к стабильности условий хроматографирования по сравнению с расчетом по высоте пика, однако само измерение площади вызывает появление новых источников ошибок. В случае узких пиков некоторые преимущества имеет измерение произведения удерживаемого объема на высоту пика. При неполном разделении пиков ошибки возрастают изза наложения и искажения контуров пиков. При работе с такими хроматограммами используют специальные приемы, опирающиеся главным образом на измерение высоты пиков.
Основными в количественной хроматографии являются методы: нормировки, нормировки с калибровочными коэффициентами, внутренней стандартизации и абсолютной калибровки.
Метод нормировки
При использовании метода нормировки сумму какихлибо параметров пиков, например сумму высот всех пиков или сумму их площадей, принимают за 100%. Тогда отношение высоты отдельного пика к сумме высот или отношение площади одного пика к сумме площадей, умноженное на 100, будет характеризовать массовую долю компонента в смеси:

где ω i – массовая доля i го компонента (в %); Пi – параметр хроматографического пика i го компонента (высота или площадь пика); n – число компонентов в анализируемой смеси.
Метод предполагает существование одинаковой зависимости величины измеряемого параметра от концентрации для всех компонентов смеси.
Метод нормировки с градуировочными коэффициентами
В методе за 100% принимается сумма параметров пиков с учетом чувствительности детектора. Различие в чувствительности детектора учитывается с помощью поправочных коэффициентов для каждого компонента. Один из преобладающих компонентов смеси считают сравнительным и поправочный коэффициент для него принимают равным единице.
Калибровочные (градуировочные) коэффициенты Ki рассчитывают по формуле:
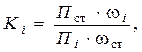
где П ст – параметр пика (высота или площадь) стандартного вещества; Пi – параметр пика определяемого компонента; ω i – массовая доля определяемого компонента; ωст – массовая доля стандарта.
За 100% принимается сумма исправленных параметров Ki·Пi. Результат анализа (массовая доля i го компонента в пробе, %) рассчитывается по формуле
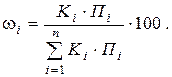 (3.14)
(3.14)
Метод абсолютной калибровки
Является наиболее точным. Экспериментально устанавливают зависимость высоты или площади пика от концентрации вещества и строят градуировочный график. Затем определяют те же характеристики пиков в анализируемой смеси и по градуировочному графику находят концентрацию вещества. Метод является основным при определении микропримесей. Он не требует разделения всех компонентов смеси, ограничиваясь лишь теми, нахождение количества которых необходимо в данном конкретном случае.
Метод внутреннего стандарта
Метод основан на введении в анализируемую смесь точного известного количества стандартного вещества. Готовят несколько (часто – пять) эталонных смесей, каждая из которых включает точно известную массу mi определяемого компонента и массу m ст стандарта. В строго одинаковых условиях хроматографируют каждую смесь и на полученных хроматограммах измеряют площади или высоты пиков Пi определяемого вещества и площадь или высоту стандарта П ст.
Поскольку величина параметра пика на хроматограмме (площадь или высота) прямо пропорциональна массе данного вещества, то
Пi = k 1 · mi, Пст = k 2 · mст,
поэтому
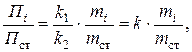 (3.15)
(3.15)
где коэффициент пропорциональности k = k 1 / k 2 или обратную ему величину 1/ k называют поправочным коэффициентом.
Затем к анализируемому раствору, содержащему неизвестную массу mx определяемого вещества, прибавляют точно известную массу стандарта m ст и хроматографируют полученный раствор в тех же условиях, что и эталонные растворы. Затем измеряют параметры Пх и П ст обоих пиков.
Иногда, наоборот, к раствору стандарта прибавляют определенное количество определяемого вещества.
По полученным данным вычисляют отношение Пх / П ст.
Окончательную обработку результатов можно проводить либо методом градуировочного графика, либо расчетным путем.
В первом случае строят градуировочный график в координатах Пх / П ст– mх / m ст, затем, зная измеренную величину Пх / П ст, по графику находят отношение mх / m сти массу mx определяемого вещества.
Во втором случае с использованием найденного поправочного коэффициента по формуле (3.15) рассчитывают отношение mх / m ст:
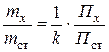 p>
p>
и, зная m ст, вычисляют массу mx определяемого вещества.
В качестве стандарта используют вещества, близкие по физикохимическим свойствам определяемому. Чем меньше различаются Пх и П ст, тем меньше ошибка определения, поэтому анализ обычно проводят в таких условиях, когда величины Пх и П ст соизмеримы. Пики стандарта и определяемого вещества не должны перекрываться.
Количественный анализ раствора «гексан – бензол – толуол» методом газожидкостной хроматографии
Цель работы: ознакомление с методом газожидкостной хроматографии; определение содержания гексана, бензола и толуола в смеси.
Сущность метода. Метод основан на разделении компонентов анализируемой смеси в результате перемещения дискретной зоны вещества вдоль слоя сорбента в потоке подвижной фазы.
В работе применяется колонка с неподвижной жидкостью карбовакс (полиэтиленгликоль), нанесенной на хроматон NAW (93% SiO2; 3,6% Al2O3; 3,4% К2О – Na2O). В качестве подвижной фазы используется азот.
После введения при помощи микрошприца анализируемой пробы в испаритель ее пары увлекаются потоком газаносителя и перемещаются по колонке, разделяясь на фракции. Скорость потока газаносителя и температура существенно влияют на хроматографию, поэтому в ходе анализа их поддерживают постоянными.
Основные узлы газо-жидкостного хроматографа ЛХМ8МД
Рассмотрим составляющие прибора (рис. 3.11) и их функции. Регулировкой редуктора на баллоне I обеспечивается заданное давление на входе хроматографа (2 атм), а регулировкой газовых потоков на блоке II – объемную скорость газаносителя. На блоке III устанавливают рабочую температуру колонки.
Программирование температуры в ходе работы должно быть выключено. На блоке IV устанавливают выбранный ток питания катарометра (чем выше ток, тем чувствительнее детектор) и задают температуру детектора, которая должна быть выше температуры кипения любого компонента разделяемой смеси на 1020 оС. На этом же блоке имеются две ручки регулировки чувствительности самописца.
Под верхней крышкой термостата V находится общий тумблер включения хроматографа, переключатель задатчика температуры испарителя, места ввода проб при помощи микрошприца в колонки 1 (ближняя) и 2 (дальняя), закрытые сменной эластичной мембраной. Здесь же находятся концевые капилляры колонок с отливами для подключения резиновой трубки пенного ротаметра, измеряющего объемную скорость газаносителя. За дверцей потенциометра VI имеется два тумблера для включения самописца и протяжки диаграммной ленты и рычаг изменения скорости редуктора (слева сверху).
Для отбора и ввода в испаритель пробы анализируемого раствора используется микрошприц.
Оборудование:
1) хроматограф ЛХМ8МД;
2) микрошприц;
3) пенный ротаметр;
4) бюксы с пробами и чистыми компонентами – 5 шт.
Реактивы:
1) бензол;
2) толуол;
3) гексан;
4) исследуемый раствор, состоящий из бензола, толуола и гексана.
Ход работы:
1. Включить прибор для прогревания в течение 0,5 ч.
2. Получить у инженера лаборатории чистые компоненты анализируемой смеси (бензол, толуол, гексан) и контрольную задачу.
3. Используя гексан, бензол и толуол, приготовить из них модельный раствор известного состава. Вычислить массовые доли компонентов в нем.
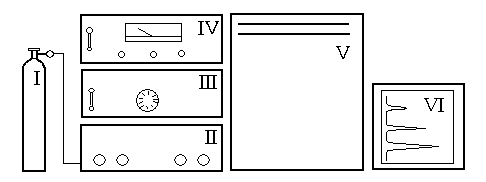
Рис. 3.11. Устройство хроматографа ЛХМ8МД:
I – баллон с газомносителем; II – блок регулировки газовых потоков; III – блок программирования температуры колонки; IV – блок управления детектором; V – блок воздушного термостата хроматографической колонки; VI – самопишущий потенциометр
4. С помощью микрошприца отобрать пробу первого раствора (или чистого вещества). Промыть микрошприц отмеряемой жидкостью, вылив первую порцию в стакан для слива. Набрать пробу еще раз.
5. Включить тумблер движения ленты самописца (рычагом редуктора должна быть задана скорость 780 мм/ч). Затем ввести иглу в испаритель первой колонки (ближнее гнездо под крышкой термостата), нажать поршень до упора и быстро вынуть иглу обратно. На ленте самописца через некоторое время появится пик прошедшего через колонку вещества. Следующую пробу можно закалывать после возвращения пера самописца на нулевую линию.
6. Получить хроматограммы веществ, перечисленных в табл. 3.6 (модельный раствор и контрольную задачу надо хроматографировать по три раза и в расчетах использовать средние величины h и w ½).
7. Провести обмер хроматограмм, как это показано на рис. 3.6. Полученные данные занести в табл. 3.6.
8. Провести обработку результатов. По величине приведенного времени удерживания tr идентифицировать на хроматограммах модельного раствора и контрольной задачи пики гексана, бензола и толуола. Измерить высоты всех пиков.
9. Используя метод нормировки с калибровочными коэффициентами, вычислить массовые доли каждого компонента в модельном растворе и в контрольной задаче:
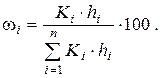
Проверить соответствие найденного количественного состава модельного раствора массовым долям компонентам, рассчитанным при его приготовлении.
10. Сделать вывод о проделанной работе.
Таблица 3.6
Объемы проб и результаты обработки хроматограмм
| Проба | Масса пробы m, мкг | Объем пробы V, см3 | ρ, мкг/см3 | tr, мм | w ½, мм | h, мм | Ki (масс.) | ω i, % |
| Гексан (Г) | 0,660 | 0,700 | –– | |||||
| Бензол (Б) | 0,879 | 0,780 | –– | |||||
| Толуол (Т) | 0,866 | 0,794 | –– | |||||
| Раствор: – гексан – бензол – толуол | … … … | … … … | 0,660 0,879 0,866 | … … … | … … … | … … … | 0,700 0,780 0,794 | … … … |
| Задача: – гексан – бензол – толуол | … … … | … … … | 0,660 0,879 0,866 | … … … | … … … | … … … | 0,700 0,780 0,794 | … … … |
Порядок работы на хроматографе ЛХМ8МД:
1. Включить шнур питания хроматографа в электрическую сеть.
2. Включить общий тумблер питания под крышкой термостата IV, а также тумблеры питания блоков II, III и самописца V (движение ленты не включать).
3. Включить на блоке III тумблер питания детектора и установить значение температуры детектора 120 oС. Тумблер полярности должен быть в положении «–».
4. Установить диском блока II температуру колонки 90 oС. После достижения заданной температуры (мигает лампочка индикатора на этом блоке) контрольным ртутным термометром, вставленным в гнездо колонки возле испарителя, уточнить и отрегулировать температуру колонки 80 oС.
5. Переключателем под крышкой термостата IV установить температуру испарителя 120 оС.
6. Открыть вентиль баллона с азотом и игольчатый вентиль на редукторе. Давление на левом манометре должно быть 2 атм. Нельзя допускать увеличение его свыше 3 атм – возможен разрыв колонки! Через час после включения хроматограф готов к работе.
7. Установить перо самописца на 0 вращением ручек «грубо» и «точно» (блок III). Во время работы возможен дрейф нулевой линии самописца. При необходимости перо повторно выводят на 0 вращением ручек «грубо» и «точно» на блоке управления детектором.p>
8. При помощи пенного ротаметра измерьте объемную скорость газаносителя V α. Если она больше чем на 5 см3/мин отличается от заданной величины – 30 см3/мин, нужна регулировка расхода газа (вентилем колонки 1 на блоке I или редуктором на баллоне). Эту работу проводить в присутствии преподавателя или инженера лаборатории.
Выключение хроматографа:
1. Выключить оба тумблера самописца V.
2. Выключить тумблеры питания блоков II и III.
3. Выключить общий тумблер питания хроматографа под крышкой термостата IV.
4. Закрыть игольчатый вентиль редуктора на баллоне с азотом.
5. Закрыть вентиль баллона.
6. Вынуть шнур питания из розетки (щитка).
7. Отодвинуть крышку испарителя колонки 1 и заменить резиновую мембрану новой (получить у инженера лаборатории).
8. Плотно задвинуть крышку испарителя.
Контрольные вопросы
1. В чем состоит принцип хроматографического разделения?
2. Какие требования предъявляют к неподвижному носителю и неподвижной жидкости в ГЖХ?
3. Абсолютные и приведенные характеристики хроматограммы.
4. Как проводят качественный анализ в газовой хроматографии? Что такое индекс удерживания Ковача?
5. Как проводят количественный анализ в газовой хроматографии? Опишите основные методы.
6. Для анализа каких объектов применяют газожидкостную хроматографию? Оцените точность и воспроизводимость ГЖХ.
Литература
1. Гольдберг, К.А. Курс газовой хроматографии / К.А. Гольдберг, М.С. Вигдергауз. М.: Химия, 1974. 375 с.
2. Столяров, Б.В. Руководство к практическим работам по газовой хроматографии / Б.В. Столяров, И.М. Савинов, А.Г. Витенберг. Л.: Химия, 1988. 336 с.
3. Вяхирев, Д.А. Руководство по газовой хроматографии / Д.А. Вяхирев, А.Ф. Шушунова. М.: Высш. школа, 1987. – 335 с.

