2015-01-30
2015-01-30 8177
8177МИНИСТЕРСТВО ОБРАЗОВАНИЯ И НАУКИ РФ
Государственное образовательное учреждение высшего профессионального образования
«НАЦИОНАЛЬНЫЙ ИССЛЕДОВАТЕЛЬСКИЙ
Томский политехнический университет»
Т.М.Гиндуллина, Н.М.Дубова
Хроматографические методы анализа
Учебно-методическое пособие
Рекомендовано к печати Редакционно-издательским советом
Томского политехнического университета
Издательство
Томского политехнического университета
УДК 543.544(076.8)
ББК 24.4я73
Г 34
Гиндуллина Т.М.
Г 34 Хроматографические методы анализа: учебно-методическое пособие /Т.М. Гиндуллина, Н.М. Дубова – Томск: Изд-во Томского политехнического университета, 2010. – 80 с.
В пособии изложены краткие сведения о теоретических представлениях в методе хроматографии, дана характеристика различных вариантов хроматографии, описаны аналитические возможности и области применения хроматографии, приведены вопросы для самоконтроля. Приведены методические указания к выполнению лабораторных работ по газожидкостной, тонкослойной, бумажной, ионообменной хроматографии. В приложении даны примеры решения типовых задач по хроматографии.
Учебно-методическое пособие соответствует рабочей программе дисциплины «Аналитическая химия и ФХМА», предназначено для студентов направления 240100– «Химическая технология и биотехнология».
УДК 543.544(076.8)
ББК 24.4я73
Рецензенты
Кандидат химических наук, доцент ТГУ
В.Н. Баталова
Кандидат химических наук, доцент СибГМУ
А.А. Блинникова
© Гиндуллина Т.М., Дубова Н.М., составление, 2010
© Составление. Томский политехнический
университет, 2010
© Оформление. Издательство Томского
политехнического университета, 2010
ГЛАВА 1. ХРОМАТОГРАФИЯ
Введение
Хроматография – важнейший аналитический метод. Хроматографическими методами можно определять газообразные, жидкие, и твердые вещества с молекулярной массой от единиц до 106. Это могут быть неорганические вещества, например, ионы металлов, изотопы водорода, и органические – белки, синтетические полимеры и т.д. С помощью хроматографии получена обширная информация о строении и свойствах органических соединений многих классов. Хроматографию с успехом применяют в исследовательских и клинических целях в различных областях биохимии и медицины, в фармацевтике, криминалистике, пищевой промышленности, для мониторинга окружающей среды. Универсальность, экспрессность, чувствительность метода обуславливают частое использование хроматографии в аналитических целях.
Возникновение хроматографии как научного метода связано с именем русского ученого-ботаника М.С.Цвета, который впервые применил явление адсорбции для анализа зеленой части хлорофилловых пигментов листьев. В 1903 г. М.С.Цвет опубликовал статью, в которой сформулировал принцип нового метода и наглядно показал возможность отделения зеленой части хлорофилловых пигментов от желтой и оранжевой с помощью углекислого кальция (адсорбента). Однако метод хроматографии не использовался вплоть до 1930 года, когда немецкие биохимики Кун, Ледерер, Винтерштейн повторили опыты Цвета и успешно разделили каротин на отдельные изомеры, предсказанные Цветом. С этого времени хроматография стала развиваться в самых разнообразных направлениях.
Первые публикации, посвященные применению метода Цвета в неорганическом анализе, относятся к 1937 году и принадлежат Швабу и его сотрудникам. В этих работах приведена методика качественного анализа смесей некоторых катионов и анионов на стеклянной колонке с оксидом алюминия. С 1938 г. широкое распространение получил метод тонкослойной хроматографии, разработанный Н.А.Измайловым и М.C.Шрайбер.
Значительные успехи в разделении и анализе неорганических веществ были достигнуты в 50-х годах, когда в практику хроматографии были введены в качестве адсорбентов ионообменные смолы, что способствовало развитию ионообменной хроматографии. В 1941 году английские ученые Мартин и Синдж предложили метод распределительной хроматографии в жидкостно-жидкостном варианте.
В 1948 г. русские ученые Е.H. Гапон и Т.Б. Гапон предложили осадочную хроматографию, основанную на различной растворимости осадков в подвижной фазе. Первая работа по газовой хроматографии в России была выполнена Н.М. Туркельтаубом в 1949г. В 1952 году Джеймс и Мартин применили газожидкостную хроматографию к анализу жирных кислот. Дальнейшему развитию газовой хроматографии способствовали работы русских ученых А.A. Жуховицкого, М.C. Вигдергауза, A.B. Киселева, Д.A. Вяхирева, А.В. Березкина и других. Более 10 работ (1957–1980), выполненных с применением хроматографических методов, были удостоены Нобелевских премий.
1. Сущность хроматографии
Хроматография – это физико-химический метод разделения веществ, основанный на распределении компонентов между двумя фазами – подвижной и неподвижной. Неподвижной фазой обычно служит твердое вещество (сорбент) или пленка жидкости, нанесенная на твердое вещество. Подвижная фаза представляет собой жидкость или газ, протекающий через неподвижную фазу.
Компоненты анализируемой смеси вместе с подвижной фазой перемещаются вдоль стационарной фазы, которую обычно помещают в колонку (стеклянную или металлическую трубку). Если молекулы разных компонентов разделяемой смеси обладают различной адсорбируемостью или растворимостью, то время их пребывания в неподвижной фазе, а следовательно, и средняя скорость передвижения по колонке различны. Одни компоненты остаются в верхнем слое сорбента, другие, с меньшей адсорбируемостью, оказываются в нижней части колонки, некоторые покидают колонку вместе с подвижной фазой. Так достигается разделение компонентов. Хроматография – динамический метод, связанный с многократным повторением сорбционных и десорбционных процессов, так как разделение происходит в потоке подвижной фазы. Это обеспечивает эффективность хроматографического метода по сравнению с методами сорбции в статических условиях.
С помощью хроматографии возможны: разделение сложных смесей органических и неорганических веществ на отдельные компоненты, очистка веществ от примесей, концентрирование веществ из сильно разбавленных растворов, качественный и количественный анализ исследуемых веществ.
2. Классификация хроматографических методов
В основу классификации многочисленных хроматографических методов положены следующие признаки:
1) агрегатное состояние фаз;
2) механизм взаимодействия сорбент – сорбат;
3) способы проведения хроматографического анализа;
4) аппаратурное оформление (техника выполнения) процесса хроматографирования;
5) цель хроматографирования.
По агрегатному состоянию фаз хроматографию разделяют на газовую и жидкостную. Газовая хроматография включает газожидкостную и газотвердофазную, жидкостная – жидкостно-жидкостную и жидкостно-твердофазную. Первое слово в названии метода характеризует агрегатное состояние подвижной фазы, второе – неподвижной.
По механизму взаимодействия сорбента и сорбата можно выделить несколько видов хроматографии: адсорбционная основана на различии в адсорбируемости веществ твердым сорбентом; распределительная основана на различной растворимости разделяемых веществ в неподвижной фазе (газожидкостная хроматография) или на различной растворимости веществ в подвижной и неподвижной фазах (жидкостная хроматография); ионообменная хроматография – на разной способности веществ к ионному обмену; эксклюзионная хроматография – на различии в размерах и формах молекул разделяемых веществ; аффинная хроматография – на специфических взаимодействиях, характерных для некоторых биологических и биохимических процессов (например, антитело и антиген, гормон и рецептор и др.). Существует осадочная хроматография, основанная на образовании отличающихся по растворимости осадков разделяемых веществ с сорбентом, адсорбционно-комплексообразовательная, основанная на образовании координационных соединений разной устойчивости в фазе или на поверхности сорбента, и др. Следует помнить, что классификация по механизму взаимодействия весьма условна: ее используют в том случае, если известен доминирующий механизм; часто процесс разделения протекает сразу по нескольким механизмам.
По технике выполнения выделяют колоночную хроматографию, когда разделение проводится в специальных колонках, и плоскостную хроматографию, когда разделение проводится на специальной бумаге (бумажная хроматография) или в тонком слое сорбента (тонкослойная хроматография). В колоночной хроматографии используют насадочные или капиллярные колонки. Насадочную колонку заполняют сорбентом (насадкой), а внутреннюю стенку капиллярной колонки покрывают пленкой жидкости или пылью адсорбента.
В зависимости от цели проведения хроматографического процесса различают аналитическую хроматографию (качественный и количественный анализ); препаративную хроматографию (для получения веществ в чистом виде, для концентрирования и выделения микропримесей); промышленную (производственную) хроматографию для автоматического управления процессом (при этом целевой продукт из колонки поступает в датчик). Хроматографию часто используют для исследовательских целей при изучении растворов, каталитических процессов, кинетики химических процессов и т.п.
Классификация по способам проведения анализа подразделяет хроматографию на три вида: 1) фронтальный, 2) проявительный, 3) вытеснительный.
Фронтальный метод наиболее прост по выполнению. Через хроматографическую колонку с сорбентом непрерывным потоком пропускают раствор или газовую смесь исследуемых веществ, сорбируемость которых увеличивается в ряду А < В < С. Соответственно этому компоненты располагаются в колонке. Однако они разделяются не полностью. В чистом виде может быть выделен лишь первый, наиболее слабо сорбирующийся компонент, который движется вдоль слоя сорбента впереди остальных. За зоной первого компонента следует в непосредственном контакте зона, содержащая первый и второй компоненты. Третья зона содержит смесь первого, второго и третьего компонентов. В некоторый момент времени сорбент насыщается, и наступает «проскок», т.е. из колонки начинают выходить компоненты в соответствии с их сорбируемостью. Если пропускать жидкость или газ, выходящие из колонки, через детектор концентраций и наносить показания его в течение всего опыта на график, то полученная выходная кривая будет иметь форму ступенчатой кривой (рис.1.1).
Фронтальный метод не нашел широкого применения в анализе, т.к. не дает полного разделения компонентов анализируемой смеси. Однако этот метод весьма эффективен для препаративного выделения чистого вещества из технического образца при условии, что это вещество удерживается в колонке слабее всех других компонентов объекта анализа.
Типичные примеры применения фронтального анализа: очистка и умягчение воды ионообменными материалами; очистка воздуха активированными углями от отравляющих веществ в противогазах и вентиляционных фильтрах химических предприятий; концентрирование ценных веществ из сточных промышленных вод металлургических предприятий; очистка лекарственных препаратов и пищевых продуктов с помощью ионообменников и т.д.
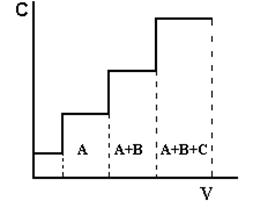 |
Рис.1.1. Выходная кривая фронтального анализа
А, В, С – разделяемые вещества
Проявительный (элюентный) метод выгодно отличается от фронтального тем, что он позволяет полностью разделить много-компонентную смесь. Хроматографическую колонку промывают растворителем или газом-носителем (элюентом), обладающим меньшей сорбируемостью, чем любое из разделяемых веществ. Затем в колонку вводят исследуемую смесь в виде порции раствора или газа, а не непрерывно, и продолжают пропускать элюент. При этом разделяемые вещества перемещаются вдоль колонки с разными скоростями в соответствии с их сорбируемостью. На выходе из колонки детектор фиксирует непрерывно концентрацию компонентов, а связанный с ним регистрирующий прибор записывает выходную кривую в виде ряда пиков, число которых соответствует числу разделенных компонентов (рис.1.2).
Проявительный метод анализа получил широкое применение как в жидкостной, так и в газовой хроматографии. Это объясняется тем, что при правильном выборе условий разделения компоненты смеси выходят из колонки в чистом виде, и их можно выделить для исследования другими методами анализа. Кроме того, качественный и количественный состав анализируемой смеси можно определить простым измерением объемов удерживания и площадей пиков соответствующих компонентов на полученной хроматограмме.
Вытеснительный метод отличается от фронтального и проявительного тем, что после введения пробы исследуемой смеси колонку
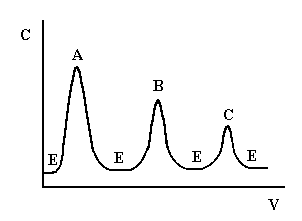 |
Рис. 1.2. Выходная кривая проявительного анализа
А, В, С – разделяемые вещества, Е – растворитель (элюент)
промывают растворителем или газом-носителем, к которым добавляют раствор вещества (вытеснитель), обладающего большей сорбируемостью, чем любое из разделяемых веществ. По мере продвижения по колонке элюент вытесняет вещество С, которое в свою очередь вытесняет вещество В и т.д. В результате вытесняемая смесь перемещается впереди фронта вытеснителя и скорость движения вещества равна скорости движения вытеснителя. Разделяемые вещества и на колонке, и в элюате располагаются последовательно друг за другом. Каждый из компонентов выделяется в чистом виде, но не количественно, так как зоны компонентов не разделены промежутками чистого сорбента.
Невозможность получения на выходе из колонки достаточно чистых компонентов разделяемой смеси, а также длительность процесса разделения затрудняют использование этого метода в аналитических целях. Однако для препаративных целей метод не потерял значения, так как возможность применения таких высокоактивных и доступных адсорбентов, как активированные угли, позволяет достигнуть высокой производительности. Достоинством метода является также то, что зоны не размываются в отличие от проявительного анализа.
Вопросы для самоконтроля
1. В чем сущность хроматографического процесса?
2. Каково назначение подвижной и неподвижной фаз?
3. Какие процессы происходят в колонке?
4. Как классифицируют методы хроматографии по агрегатному состоянию фаз и по способу хроматографирования??
5. В чем состоит проявительный (элюентный) анализ?
6. В чем преимущество элюентной хроматографии перед фронтальной и вытеснительной?
7. Как классифицируют методы хроматографии по технике проведения эксперимента и цели?
8. В чем сущность хроматографического разделения по методу: а) газожидкостной хроматографии; б) распределительной жидкостной хроматографии; в) осадочной хроматографии; г) тонкослойной хроматографии; д) ионообменной хроматографии; е) эксклюзионной хроматографии?
9. Как влияет температура на хроматографический процесс?
3. Ионообменная хроматография
В основе ионообменной хроматографии лежит обратимый стехиометрический обмен ионов, содержащихся в хроматографируемом растворе, на ионы веществ, называемых ионитами или ионобменниками. Иониты могут быть органические и неорганические, природные и синтетические. По знаку обменивающихся ионов различают катиониты (для обмена катионов) и аниониты (для обмена анионов).
К природным ионитам относятся алюмосиликаты, некоторые сорта каменных углей, мягкие и твердые угли даже без предварительной обработки.
В аналитической практике широко используют синтетические иониты. Ионообменники получают реакциями поликонденсации либо полимеризации, линейные цепи полимеров разветвлены и связаны друг с другом «мостиками», например, молекулами дивинилбензола; в состав ионитов входят различные функциональные (ионогенные) группы, которые и определяют наиболее характерные свойства ионитов. Иониты нерастворимы в воде, кислотах, щелочах и во многих органических растворителях, но способны набухать в воде за счет гидрофильных ионогенных групп.
Органические катиониты содержат кислотные функциональные группы: – SO3–, – PO3–, – COO–, – OH –. Органические аниониты содержат группы основного характера: – NH2+,= NH+,  N+, – N(CH3)3+. Катиониты представляют собой полиэлектролиты, диссоциирующие с образованием высокомолекулярного аниона (например, RSO3–) и подвижного катиона (например, Н+- иона),легко обменивающегося на другие катионы. Аниониты диссоциируют на высокомолекулярный катион (например, RNH+) и подвижный анион (например, ОН–), способный обмениваться на другие анионы (R – высокомолекулярный углеводородный радикал ионообменной смолы).
N+, – N(CH3)3+. Катиониты представляют собой полиэлектролиты, диссоциирующие с образованием высокомолекулярного аниона (например, RSO3–) и подвижного катиона (например, Н+- иона),легко обменивающегося на другие катионы. Аниониты диссоциируют на высокомолекулярный катион (например, RNH+) и подвижный анион (например, ОН–), способный обмениваться на другие анионы (R – высокомолекулярный углеводородный радикал ионообменной смолы).
Реакции ионного обмена можно представить схематично сле-дующим образом:
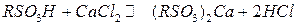
(катионный обмен)
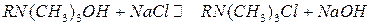
(анионный обмен)
Реакции ионного обмена обратимы и в первом приближении подчиняются закону действующих масс.
Важной характеристикой ионита является его обменная емкость.
3.1. Обменная емкость ионитов
Обменная емкость (ОЕ) – количественная мера способности ионита поглощать противоионы. Численно обменную емкость выражают количеством поглощенных миллимоль эквивалентов ионов на 1г сухой смолы в Н+-форме для катионита и Сl --форме для анионита.
Определение емкости можно отнести и к единице объема набухшего слоя ионита. Обменная емкость, полученная в статических условиях, когда навеску ионита помещают в раствор насыщающего иона определенной концентрации и выдерживают при встряхивании до полного насыщения ионита, называется статической (СОЕ). Величина ее отличается от величины обменной емкости, полученной в динамических условиях при пропускании насыщающего раствора через колонку с ионитом.
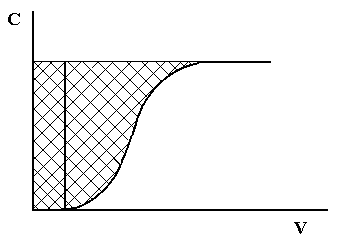 |

Рис.1.3. Выходная хроматографическая кривая
Динамическая обменная емкость характеризуется двумя показателями: динамической обменной емкостью до проскока (ДОЕ) и полной динамической емкостью (ПДОЕ). ДОЕ представляет собой емкость ионита, определяемую по появлению данного иона в вытекающем из колонки растворе. ПДОЕ определяется по полному прекращению извлечения данного иона из раствора. Это различие можно пояснить графически на рисунке 1.3.
ДОЕ определяется площадью прямоугольника, основанием которого является объем раствора, вытекающего из колонки до наступления проскока иона, а высотой – исходная концентрация обменивающегося иона. ПДОЕ выражается площадью над выходной хроматографической кривой.
ДОЕ всегда меньше, чем полная динамическая обменная емкость, и зависит от ряда факторов: от типа ионита, состава раствора, размера зерен ионита и скорости протекания раствора.
3.2. Классификация ионитов
От вида функциональных групп, входящих в состав ионита, зависит, насколько сильно выражены кислотные или основные его свойства. В зависимости от этого различают четыре группы ионитов.
1. Сильнокислотные катиониты имеют в качестве функциональных групп сульфогруппу –SO3-и фосфорную группу –РО3-. Они используются в кислых, нейтральных и щелочных средах. Это сульфокислотные катиониты полистирольного типа марок КУ-2, КУ-23, СДВ, СБС. К фосфорнокислым относятся катиониты марок КФ-2, КФ-11.Катиониты полистирольного типа выпускаются в виде сферических гранул и имеют либо янтарную, либо светло-желтую окраску.
Катиониты фенольного типа, например, КУ-1, окрашены в черный цвет, их частицы имеют неправильную форму. Такие катиониты бифункциональны, т.е. наряду с группой –SO3-имеют в своем составе группу –ОН-.Преимущество полистирольных катионитов – их монофункциональность, высокая обменная емкость, высокая термическая устойчивость.
2. Слабокислотные катиониты имеют в качестве функцио-нальных групп карбоксильные группы – СОО-, – ОН-. Это катиониты марок КБ-1, КБ-4, КФУ-1. Катиониты с карбоксильными группами окрашены в белый или светло-зеленый цвет. Важным свойством подобных катионитов является их высокое сродство к иону водорода. Даже небольшого количества разбавленной соляной кислоты достаточно для полной регенерации катионита. Слабокислотные катиониты работают в щелочных и нейтральных средах.
3. Сильноосновные (высокоосновные) аниониты имеют в качестве функциональных групп четвертичные аммониевые группы. Это аниониты марок АВ-16, АВ-17, АВ-18, АВ-20. Они могут применяться для хроматографирования в кислых, щелочных и нейтральных средах. Сильноосновные аниониты имеют желтую или светло-желтую окраску. Они часто используются для разделения большинства ионов металлов. Ион щелочных, щелочноземельных, редкоземельных элементов, алюминия, никеля, меди и др. не сорбируются анионитами при любой концентрации соляной кислоты. Остальные ионы металлов в пределах концентрации НСl от 0,1 до 12 моль/л сорбируются анионитами в различной степени, т.к. образуют анионные хлоркомплексы, имеющие сильно отличающиеся константы нестойкости.
4. Слабоосновные (низкоосновные) аниониты в качестве функциональных групп имеют аминогруппы разной степени замещения:
– NH2+,= NH+, N+. Это аниониты марок АН-2Ф,АН-1, АН-23 и др.Они работают в кислых и нейтральных средах. Анионит ЭДЭ-10П содержит несколько активных аминогрупп вторичного, третичного и четвертичного аммониевых оснований. Поэтому он обладает и слабоосновными, и в некоторой степени сильноосновными свойствами.
3.3. Практическое применение ионообменной хроматографии
Методы ионообменной хроматографии используют преимущественно для разделения ионов. Простейшая методика разделения заключается в поглощении смеси компонентов и последовательном элюировании каждого компонента подходящим растворителем. Иониты используют также в водоподготовке (умягчение воды, опреснение морской воды); в гидрометаллургии и гальванотехнике (селективное извлечение ценных металлов из производственных растворов и сточных вод; в пищевой и гидролизной промышленности (очистка сахаросодержащих растворов, осветление плодово-ягодных соков и т.д.); в медицине и фармацевтической промышленности (очистка лекарственных препаратов, антибиотиков).
Рассмотренные области применения ионообменных смол не исчерпывают всего многообразия, однако они показывают широкие возможности, которые открывают использование ионитов в аналитической химии и технологии.
Вопросы для самоконтроля
1. В чем сущность метода ионообменной хроматографии?
2. Как подготовить ионообменную смолу к работе?
3. Какие функциональные группы обеспечивают обменные свойства различных синтетических ионообменных смол? Какие типы катионитов и анионитов Вам известны?
4. Что такое «обменная емкость» ионита, в каких единицах измеряется?
5. Как определяют: а) статическую обменную емкость ионита; б) динамическую обменную емкость ионита?
6. Зависит ли селективность ионообменника от его емкости?
7. Как провести деионизацию воды с помощью ионообменников? Напишите уравнения реакций.
8. Каковы области применения, достоинства и недостатки ионообменной хроматографии?
К плоскостным видам хроматографии относят бумажную (БХ) и тонкослойную (ТСХ). Эти два вида жидкостной хроматографии просты по технике выполнения, экспрессны, не требуют дорогостоящего оборудования. Разделение этими методами может быть выполнено с использованием хроматографических систем жидкость–твердый сорбент и жидкость–жидкость–твердый сорбент, поэтому выделяют адсорбционную, распределительную, обращенно-фазовую и ионообменную плоскостную хроматографию. Тонкослойную хроматографию используют чаще, чем бумажную.
4.1. Тонкослойная хроматография
Метод тонкослойной хроматографии был разработан Н. А. Измайловым и М. С. Шрайбер еще в 1938 г. В методе ТСХ неподвижная твердая фаза (силикагель, оксид алюминия, порошок целлюлозы) тонким слоем наносится на стеклянную, пластмассовую или металлическую пластинку. В качестве подвижной фазы используют различные растворители или их смеси, органические и неорганические кислоты. Выбор растворителя зависит от природы сорбента и свойств анализируемых соединений. Например, при хроматографировании аминокислот используют смесь n-бутанола с уксусной кислотой и водой, при анализе неорганических ионов – водные буферные растворы, создающие постоянное значение рН. В ТСХ чаще используют восходящий способ получения хроматограммы. Раствор образца наносят микропипеткой на небольшом расстоянии от края пластинки на стартовую линию, и край пластинки погружают в растворитель, который действует как подвижная фаза жидкостной адсорбционной хроматографии. Под действием капиллярных сил растворитель поднимается вверх по пластинке и с разной скоростью переносит за собой компоненты смеси, что приводит к их пространственному разделению. Чтобы растворитель не испарялся с поверхности сорбента, пластинка на время разделения должна быть помещена в герметически закрытую прозрачную камеру. Разделяемые компоненты на пластинке образуют отдельные зоны (пятна). Хроматографирование продолжают до тех пор, пока растворитель не пройдет от линии старта около 10 см до так называемой линии фронта. После этого пластинку вынимают из хроматографической камеры, подсушивают на воздухе и определяют положение пятен.
В нисходящей хроматографии растворитель передвигается по слою вниз под действием и капиллярных, и гравитационных сил. Горизонтальная хроматография выполняется в виде круговой и со свободным испарением растворителя. В круговой хроматографии в центр горизонтально установленной пластинки вносят каплю анализируемой смеси и непрерывно подают растворитель, который под действием капиллярных сил движется в радиальном направлении от центра. Компоненты смеси располагаются в слое в виде концентрических колец.
Схема разделения смеси веществ методом тонкослойной хроматографии приведена на рис.1.4. Пятна характеризуют положение компонентов А, В, С на пластинке в конце опыта.
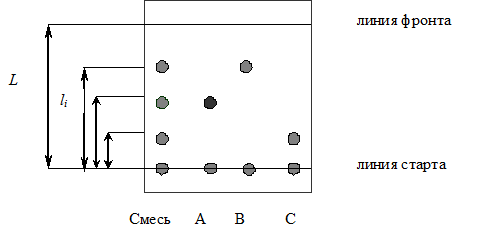
Рис.1.4. Схема разделения методом восходящей тонкослойной хроматографии
Сорбционные свойства системы в ТСХ характеризуются под-вижностью Rf – относительной скоростью перемещения компонентов в тонком слое. Величины Rf рассчитываются из экспериментальных данных (рис.1.4.):
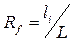 (1.1),
(1.1),
где li – расстояние от стартовой линии до центра пятна, L – расстояние, пройденное растворителем от стартовой линии до границы фронта растворителя.
Rf характеризует положение пятна на хроматограмме. Это константа для данного вещества на данном сорбенте в данной системе растворителей. На величину Rf влияют качество и активность сорбента, его влажность, толщина слоя, качество и природа растворителя, техника эксперимента (способ нанесения пробы, способ детектирования) и другие факторы. На практике часто пользуются относительной величиной
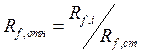 (1.2)
(1.2)
Где Rf,ст также рассчитывают по уравнению (1.1).
Разделение двух веществ с Rf,1 и Rf,2 практически возможно, если Rf,1> Rf,2 и Δ Rf ≥ 0,1. Эффективность выбранного варианта ТСХ (адсорбционного, распределительного, ионообменного) и хроматографической системы можно оценить по фактору разделения (селективности) двух веществ с разными коэффициентами распределения:
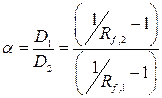 (1.3)
(1.3)
Качественный анализ. Проще всего идентификация вещества может быть сделана, если пятно определяемого вещества имеет характерную окраску. Невидимые хроматограммы проявляют соответствующими реагентами, как правило, групповыми. По характерной окраске образующихся цветных зон судят о составе анализируемой пробы. При обработке пластинки, например, парами иода четко проявляются непредельные соединения; при опрыскивании пластинки тиоцианатом кобальта амины образуют голубые пятна на розово-белом фоне. В физических методах проявления используется способность некоторых веществ флуоресцировать под действием УФ-излучения.
Наиболее общий подход к качественному анализу основан на значениях Rf. При соблюдении стандартных условий получаются воспроизводимые значения Rf, которые можно использовать в аналитических целях при сравнении с табличными, если они получены в тех же условиях опыта; более надежно использовать значения Rf,отн.
Самым надежным способом является метод свидетелей (стандартных веществ). Стандартное вещество в том же растворителе наносится на стартовую линию рядом с анализируемой пробой и, таким образом, хроматографируется в тех же условиях (рис.1.4).
Таблица 1.1
Подвижные фазы, проявители, величины Rf некоторых катионов при разделении на микрокристаллической целлюлозе методом ТСХ
| Катион | Подвижная фаза, % | Проявитель | Rf |
| Hg(I) | н-бутанол–вода (85:15); рН 3,0 (СН3СООН) | Водный раствор К2CrO4 | 0,13 |
| Ag(I) | 0,11 | ||
| Pb(II) | 0,05 | ||
| Zn(II) | Этанол–5 М HCl(90:10) | Дитизон | 0,93 |
| Fe(III) | Самоидентификация | 0,80 | |
| Co(II) | 1-Нитрозо-2-нафтол | 0,33 | |
| Ni(II) | Диметилглиоксим | 0,33 | |
| Ca(II) | Изопропанол–вода– 1 М HCl (40:20:20) | Ализарин | 0,73 |
| Sr(II) | Родизонат калия | 0,66 | |
| Ba(II) | Родизонат калия | 0,55 |
По окончании хроматографирования и проявления хроматограммы приступают к идентификации веществ. Совпадение Rf компонента пробы и одного из свидетелей дает основание для отождествления веществ.
Количественные определения в ТСХ могут быть сделаны непосредственно на пластинке, в этом случае каким-либо способом измеряют площадь пятна и по заранее построенному градуировочному графику находят количество вещества. Применяется также прямое спектрофотометрирование пластинки по спектрам отражения и по спектрам поглощения (фотоденситометрия), для количественных расчетов предварительно строят градуировочный график, используя оптическую плотность в центре пятна Наиболее точным считается метод, когда анализируемое вещество удаляют с пластинки механическим путем или вымывают подходящим растворителем после вырезания зоны, а затем анализируют спектрофотометрическим, флуориметрическим, атомно-абсорбционным методами.
Метод ТСХ прост по методике выполнения и аппаратуре, экспрессен и не требует для анализа больших количеств вещества. Метод широко используется для идентификации компонентов лекарств, биохимических препаратов, неорганических веществ.
4.2. Бумажная хроматография
Вместо пластинок с нанесенным тонким слоем сорбента можно использовать специальную хроматографическую бумагу в виде листов или полосок. Хроматографическая бумага должна быть химически чистой, нейтральной, инертной по отношению к компонентам раствора и подвижной фазе и быть однородной по плотности; имеют значение структура молекул целлюлозы в бумаге, ориентация волокон и другие свойства, влияющие на скорость движения подвижной фазы. Основные операции в бумажной хроматографии проводятся примерно так же, как и в тонкослойной.
Для разделения водорастворимых веществ, например, неорганических ионов, в качестве подвижной фазы обычно берут органический растворитель, а в качестве неподвижной – воду (бумагу заранее смачивают водой). Для разделения компонентов, хорошо растворимых в органических растворителях, гидрофильную бумагу превращают в гидрофобную, пропитывая ее растворами органических веществ (парафина, растительного масла и др.), а в качестве подвижной фазы используют воду, водный раствор какой-либо кислоты или щелочи, буферный раствор.
Растворители подвижной и неподвижной фаз не должны смешиваться, состав растворителя в процессе хроматографирования не должен изменяться, растворители должны легко удаляться с бумаги. Индивидуальные растворители используются достаточно редко. Чаще для этой цели применяют смеси веществ, например, бутилового или амилового спирта с метиловым или этиловым, смеси бутилового спирта с уксусной кислотой, аммиаком и др.
По технике выполнения различают следующие виды бумажной хроматографии: одномерную, двумерную, круговую и электрофоретическую. Для получения двумерных хроматограмм хроматографирование проводят дважды: после обработки пробы одним растворителем хроматограмму поворачивают на 90° и хроматографируют вторично уже другим растворителем. Такая методика позволяет проводить более тонкие разделения компонентов смеси. Специфическим приемом является сочетание БХ и электрофореза. Для этого к влажному листу хроматографической бумаги прикладывают постоянное электрическое напряжение. Дополнительное воздействие электрического поля приводит к более четкому разделению, особенно для ионов с разными зарядами. Электрофорез можно проводить одновременно с хроматографированием, а также до или после хроматографирования.
Качественный состав пробы в методе бумажной распределительной хроматографии так же, как и в ТСХ, может быть установлен или по специфической окраске отдельных пятен на хроматограмме, или по численному значению Rf каждого компонента. Количественные определения в БХ выполняются по хроматографическим характеристикам (по площади пятна на хроматограмме и интенсивности его окраски) или после вымывания подходящим физико-химическим методом. Отметим, что метод бумажной хроматографии, предложенный в 1941 г. Мартином и Синджем, в настоящее время используют в аналитических лабораториях довольно редко.
Вопросы для самоконтроля
1. Каковы преимущества двумерной хроматографии перед одномерной бумажной или ТСХ?
2. Как идентифицировать пятна органических соединений в методе ТСХ?
3. Как выполняют количественный анализ в методе ТСХ?
4. Как определяют Rf в методе БХ и ТСХ? От чего зависит величина Rf и какие условия нужно поддерживать постоянными при проведении эксперимента?
5. Как можно определить концентрации компонентов смеси после разделения методом БХ или ТСХ?
6. Как выполняется качественный анализ с помощью плоскостных вариантов хроматографии – БХ и ТСХ?
7. Какими способами проба анализируемой смеси веществ вводится в хроматографическую установку в бумажной хроматографии?
8. Почему в методе ТСХ необходимо герметически закрывать камеру с растворителем и пластинкой во время подъема фронта растворителя?
9. Как обнаруживают и идентифицируют компоненты на бумажных и тонкослойных хроматограммах?
10. Каковы области применения, достоинства и недостатки тонкослойной хроматографии?
Газовая хроматография – это вариант хроматографии, в котором подвижной фазой является инертный газ (газ-носитель), протекающий через неподвижную фазу, обладающую большой поверхностью. Обычно в качестве подвижной фазы используют гелий, азот, аргон, водород, диоксид углерода или воздух. Газ-носитель должен быть инертным по отношению к разделяемым веществам и сорбенту, взрывобезопасным и достаточно чистым. Выбор газа-носителя в каждом конкретном случае должен обеспечивать соответствие его физических свойств получению высокой эффективности колонки и достаточной чувствительности детектора.
В зависимости от агрегатного состояния неподвижной фазы газовая хроматография подразделяется на газоадсорбционную, когда неподвижной фазой является твердый адсорбент, и газожидкостную, когда неподвижной фазой является жидкость, нанесенная на поверхность твердого носителя. В газовой хроматографии используется преимущественно элюентный (проявительный)способ проведения процесса хроматографирования.
Газовая хроматография – метод разделения летучих соединений. Этим методом можно проанализировать газообразные, жидкие и твердые вещества с молекулярной массой меньше 400, удовлетворяющие определенным требованиям, главные из которых – летучесть, термостабильность, инертность и легкость получения. Количественный анализ можно провести только в том случае, если вещество термостойко, т.е. испаряется в дозаторе воспроизводимо и элюируется из колонки без разложения. При разложении вещества на хроматограмме появляются ложные пики, относящиеся к продуктам разложения. Вещество не должно образовывать устойчивых сольватов при растворении в неподвижной жидкой фазе и реагировать с материалами, из которых изготовлены детали хроматографа. Этим требованиям удовлетворяют, как правило, органические вещества, поэтому ГХ чаще используют как метод анализа органических соединений, хотя этим методом можно определять почти все элементы периодической системы в виде летучих соединений.
5.1. Газотвердофазная хроматорафия
Вгазоадсорбционной хроматографии в качестве неподвижной фазы применяют различные адсорбенты – высокодисперсные искусственные или природные тела с высокой удельной поверхностью (10–1000 м2/г), поглощающие газы или пары. Адсорбция молекул из газовой фазы происходит за счет межмолекулярных взаимодействий, имеющих электростатическую природу; возможно образование водородной связи, но вклад этого взаимодействия уменьшается с ростом температуры.
Адсорбент должен обладать следующими основными свойствами: необходимой селективностью, отсутствием каталитической активности и химической инертностью к компонентам разделяемой смеси, достаточной механической прочностью.
Основными адсорбентами, применяемыми в газо-адсорбционной хроматографии, являются активированные угли, силикагели, оксид алюминия. Неоднородность поверхности активных адсорбентов не дает возможности определять сильно адсорбирующиеся полярные молекулы, однако, в последнее время промышленностью выпускаются адсорбенты с достаточно однородной поверхностью, такие, как пористые стекла, пористые полимеры, синтетические цеолиты (молекулярные сита), макропористые силикагели (силохром, порасил, сферосил), позволяющие проводить анализ смесей сильнополярных веществ.
Наиболее широко метод газоадсорбционной хроматографии применяют для анализа смесей газов и низкокипящих углеводородов, не содержащих активных функциональных групп. Например, для разделения О2, N2, СО, СН4, СО2 с успехом применяют глинистые материалы, сорбенты, называемые порапаками, используют для разделения гидридов металлов (Ge, As, Sn, Sb). Метод ГАХ на колонках с пористыми полимерными сорбентами– удобный и быстрый способ определения воды в неорганических и органических материалах.
5.2. Газожидкостная хроматография
В аналитической практике чаще используют метод газожидкостной хроматографии. Это связано с чрезвычайным разнообразием жидких неподвижных фаз. Вгазожидкостнойхроматографии неподвижной фазой служит практически нелетучая при температуре колонки жидкость, нанесенная на твердый носитель. Количество жидкой фазы составляет 5-30% от массы твердого носителя.
К жидкой фазе предъявляется ряд жестких требований:
1) способность хорошо растворять компоненты смеси (если растворимость мала, компоненты выходят из колонки очень быстро); 2) инертность по отношению к компонентам смеси и твердому носителю; 3) малая летучесть (чтобы не испарялась при рабочей температуре колонки); 4) термическая устойчивость; 5) достаточно высокая селективность, т.е. способность разделять смесь компонентов; 6) небольшая вязкость (иначе замедляется процесс диффузии); 7)способность образовывать при нанесении на носитель равномерную пленку, прочно с ним связанную.
Природа жидкой фазы является тем основным фактором, который определяет последовательность выхода компонентов из колонки. В качестве жидких фаз применяются неполярные парафины (например, сквалан, вазелиновое масло, апиезоны), умеренно полярные (сложные эфиры, нитрилы и др.) и полярные (полиэтиленгликоли или карбоваксы, гидроксиламины и др.)
Каждая жидкая фаза имеет температурные пределы применения. Нижний температурный предел – минимальная рабочая температура, соответствующая застыванию жидкой фазы. Обычно выбирают минимальную рабочую температуру колонки выше точки застывания жидкой фазы на 10-15оС. Верхний температурный предел – максимальная допустимая рабочая температура (МДРТ) жидкой фазы, выше которой она начинает разрушаться, при этом образуются летучие соединения, уносимые из колонки. Практика использования жидких фаз для анализа показывает, что необходимо работать с ними при температурах на 20- 30оС ниже МДРТ жидкой фазы.
Наибольшим температурным диапазоном использования в газо-жидкостной хроматографии обладают кремнийорганические полимеры, например, метилсиликоны – жидкости при комнатной температуре, а МДРТ их достигает 300-350оС. Наиболее термостабильными жидкими фазами являются карборан-силоксановые полимеры, в которые входят атомы бора, кремния и углерода. МДРТ этих соединений достигает400оС.
Твердым носителем обычно служит практически инертное твердое вещество, на которое наносят неподвижную жидкость. Основное назначение твердого носителя в хроматографической колонке – удерживать жидкую фазу на своей поверхности в виде однородной пленки. В связи с этим твердый носитель должен иметь значительную удельную поверхность (0,5-10 м2/г), причем она должна быть макропористой во избежание адсорбции компонентов пробы. Кроме того, твердый носитель должен обладать следующими качествами: отсутствием каталитической активности, достаточной механической прочностью, стабильностью при повышенных температурах, однородностью пор по размерам, максимальной однородностью размера зерен. Однако до настоящего времени не создано универсального носителя, удовлетворяющего всем перечисленным требованиям.
В качестве твердых носителей в газо-жидкостной хроматографии используются диатомиты (кизельгур, инфузорная земля), синтетические кремнеземы (макропористые силикагели, широкопористые стекла, аэросилогели), полимерные носители на основе политетрафторэтилена и т.д. Часто используют модифицированные носители, ковалентно связанные с «жидкой» фазой. При этом стационарная жидкая фаза более прочно удерживается на поверхности даже при самых высоких температурах колонки. Химически связанная неподвижная фаза более эффективна.
5.3. Аппаратурное оформление газовой хроматографии
Для проведения газо-хроматографических анализов применяются специальные приборы – газовые хроматографы.
Газовые аналитические (лабораторные) хроматографы предназначены для разделения и анализа исследуемых смесей. Это хроматографы марок ХЛ-3, ЛХМ-8МД, ЛХМ-80, модели лабораторных хроматографов, объединенных общим названием «Цвет-100». В настоящее время разработаны аналитические газовые хроматографы серии «Цвет- 500», «Цвет-500М», «Цвет-2000», «Милихром АО2».
Кроме аналитических имеются промышленные хроматографы двух типов: автоматические – для контроля производственных процессов (ХТП-63, ХПА-4, ХП-499) и препаративные – для получения чистых веществ (Эталон-1).
Промышленные газовые хроматографы отличаются от лабораторных устройством для автоматического ввода пробы, а также наличием устройства-преобразователя выходного сигнала прибора в форму, удобную для представления оператору. Промышленные хроматографы выполняются в виде двух самостоятельных блоков, один из которых устанавливается в производственном помещении вблизи точки отбора пробы. Второй блок может быть размещен на большом расстоянии от первого на пульте контрольно-измерительных приборов.
Промышленные хроматографы применяются для контроля процессов выделения и очистки (например, в производстве легких бензинов, синтетического каучука, этилового спирта), для контроля реакционных процессов, таких как полимеризация, пиролиз, синтез разнообразных продуктов (например, синтез формалина, аммиака, окиси этилена), для контроля токсических веществ в воздухе промышленных предприятий и т.д.
В настоящее время промышленные газовые хроматографы получили всеобщее признание как основное техническое средство контроля и регулирования технологических процессов химических и нефтехимических предприятий.
5.3.1.Основные узлы газового хроматографа
Современный газовый хроматограф состоит из следующих основных частей (рис.1.5):
1. Устройство подготовки пробы для хроматографического анализа (обогащение, концентрирование, пиролиз).
2. Баллон с газом-носителем и блок подготовки газа-носителя, включающий в себя очистку газа, установку расхода газа или давления, измерение расхода газа.
3. Устройство для ввода пробы и для ее испарения – дозатор-испаритель.
4. Блок анализатора, включающий в себя хроматографическую колонку и термостат колонки, регулирующий нужную температуру и измеряющий ее.
5. Детектор, преобразующий изменение состава компонентов в электрический сигнал.
6. Регистратор, записывающий результаты хроматографического анализа.
7. Электронный интегратор, автоматически фиксирующий площадь пика и время его выхода; цифропечатающее устройство, дисплей.
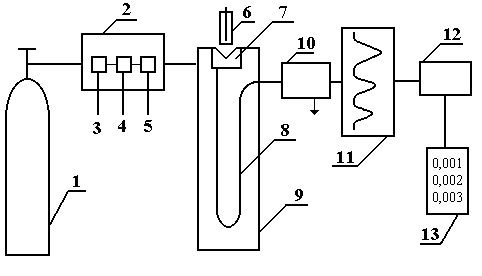 |
Рис. 1.5. Блок-схема газового хроматографа
1 – баллон со сжатым газом; 2 – блок подготовки газа-носителя; 3 – регулятор расхода газа; 4 – измеритель расхода газа; 5 – фильтр; 6 – микрошприц для введения пробы; 7 – испаритель; 8 – хроматографическая колонка; 9 – термостат; 10 – детектор; 11– самописец; 12 – интегратор; 13 – цифропечатающее устройство
Одним из основных узлов газового хроматографа является дозатор, который предназначен для точного количественного отбора пробы и введения ее в хроматографическую колонку. В каждом хроматографе дозатор-испаритель устанавливается непосредственно у входа в хроматографическую колонку. Он представляет собой небольшую емкость, соединенную с началом хроматографической колонки и снабженную самоуплотняющейся термостойкой резиновой мембраной.
В дозаторе следует поддерживать такую температуру, при которой происходило бы полное и быстрое испарение жидкого образца. Жидкую пробу дозируют микрошприцем, впуск газообразных проб часто осуществляют медицинским шприцем. В зависимости от концентрации и числа разделяемых компонентов объем вводимого газообразного образца колеблется от 1 до 10 мл, а объем жидкого образца – от 0,1 до 10 мкл.
Вместе с газом-носителем введенный парообразный образец поступает в колонку, где происходит его сорбция.
Хроматографические колонки различны по форме, размерам и материалам. Наиболее распространены спиральные, U- и W - образные колонки длиной от 2 ми менее до нескольких десятков метров. Внутренний диаметр колонок обычно от 3 до 6 мм. Колонки изготавливают из нержавеющей стали, меди, латуни, стекла. Материал колонок должен обладать химической инертностью по отношению к компонентам пробы.
Большое влияние на сорбируемость газа оказывает температура, поэтому хроматографические колонки, как правило, термостатируются. Обычно термостатирование производится при температурах, значительно превышающих комнатные, однако в некоторых случаях создаются температуры ниже 0оС при разделении низкокипящих газов.
Для обнаружения изменений в составе газа, прошедшего через колонку, предназначен детектор. Последний непрерывно измеряет концентрацию компонентов на выходе их из хроматографической колонки и преобразует концентрацию в электрический сигнал, который регистрируется самопишущим прибором.
5.3.2. Детекторы
Одним из наиболее распространенных детекторов является катарометр. Принцип его работы основан на измерении сопротивления нагретой вольфрамовой нити, которое зависит от теплопроводности омывающего газа. Количество теплоты, отводимое от нагретой нити при постоянных условиях, зависит от состава газа. Чем больше теплопроводность газа-носителя, тем большей чувствительностью будет обладать катарометр. Наиболее подходящим газом-носителем с этой точки зрения является водород, теплопроводность которого значительно превышает соответствующую характеристику большинства других газов. Однако в целях техники безопасности чаще применяется гелий, теплопроводность которого также достаточно велика. Достоинствами катарометра являются простота, достаточная точность и надежность в работе. Однако из-за невысокой чувствительности он не применяется для определения микропримесей.
Наибольшей чувствительностью обладают ионизационные детекторы, например, пламенно-ионизационный, позволяющий обнаруживать до 10-12 г. В этих детекторах измеряют электрическую проводимость пламени водородной горелки. При появлении в пламени водорода примесей органических соединений происходит ионизация пламени, пропорциональная концентрации примеси, что легко может быть измерено. Недостатком данного детектора является то, что он применим только для анализа органических веществ, а к неорганическим, таким как аммиак, сероводород, кислород, азот, оксид серы, оксид углерода и т.д., чувствительность детектора резко падает.
Таблица 1.2
Сравнительные характеристики хроматографических детекторов
| Детектор | Предел обнаружения | Диапазон линейности детектора |
| Катарометр | 10–12 г/мл | 105 |
| Пламенно-ионизационный | 10–12 г/с | 107 |
| Электронного захвата | 10–14 г/мл | 104 |
| Термоионный | 10–15 г/с | 103 |
| ИК-спектрометр | >1 мкг | 103 |
| Масс-спектрометр | 10–14 – 10–12 г | 104 |
Очень высокой чувствительностью обладает аргонный детектор, ионизация в котором происходит при столкновении молекул определяемого вещества с метастабильными атомами аргона, образующимися под действием радиоактивного b-излучения.
В термоионном детекторе в пламя горелки вводят соли щелочных металлов. При попадании в такое пламя соединений фосфора появляется ионный ток, пропорциональный содержанию атомов фосфора. Это селективный фосфорный детектор высокой чувствительности.
Известны другие типы детекторов: термохимические, пламенно-фотометрические, микрокулонометрические, ультразвуковые и т.д.
Вопросы для самоконтроля
1. Какова роль подвижной фазы в газовой хроматографии?
2. Какими способами проба анализируемой смеси веществ вводится в хроматографическую установку в газовой хроматографии?
3. Какое практическое значение имеет газовая хроматография?
4. Каковы области применения, достоинства и недостатки методов адсорбционной хроматографии?
5. Какие требования предъявляются к адсорбентам и растворителям? Какие устройства используют в качестве дозаторов?
6. Какие требования предъявляются к жидкой фазе в газожидкостной хроматографии? Какие вещества используют в качестве жидкой фазы? В качестве твердого носителя?
7. В каком хроматографическом методе основной фактор, определяющий удерживание компонента – растворение в неподвижной фазе?
8. Назовите три способы детектирования в газовой хроматографии.
9. Какова роль основных узлов в газовом хроматографе?
5.4. Характеристики удерживания
Если поток газа-носителя, содержащий десорбированное ве-щество, проходит через чувствительный элемент прибора, фикси-рующего мгновенное изменение концентрации вещества в газе (детектор), то на записывающем устройстве этого прибора получается кривая, называемая хроматографическим пиком или кривой элюирования.
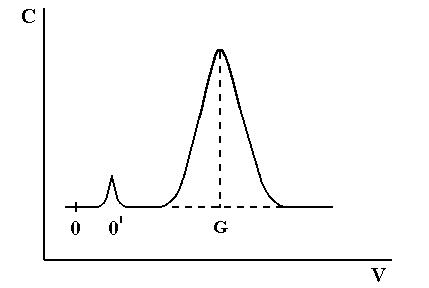 |
На рисунке 1.6. изображена типичная кривая элюирования. По оси абсцисс отложен объем элюата (можно отложить время хроматографирования). Ее параметры, называемые характеристиками удерживания, могут служить средством идентификации компонентов разделяемой смеси.
Рис.1.6. Кривая проявительного анализа (хроматографический пик)
Время от момента ввода анализируемой пробы до регистрации максимума пика называют временем удерживания (элюирования) 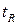 .(отрезок OG на графике). Время удерживания складывается из двух составляющих – времени пребывания вещества в подвижной и неподвижной фазах. Первое фактически равно времени прохождения через колонку несорбируемого компонента (отрезок ОО/ на графике). Значение не зависит от количества пробы, но зависит от природы вещества и сорбента, скорости потока газа-носителя, а также упаковки сорбента и может меняться от колонки к колонке. Поэтому для характеристики истинной удерживающей способности следует ввести исправленное время удерживания
.(отрезок OG на графике). Время удерживания складывается из двух составляющих – времени пребывания вещества в подвижной и неподвижной фазах. Первое фактически равно времени прохождения через колонку несорбируемого компонента (отрезок ОО/ на графике). Значение не зависит от количества пробы, но зависит от природы вещества и сорбента, скорости потока газа-носителя, а также упаковки сорбента и может меняться от колонки к колонке. Поэтому для характеристики истинной удерживающей способности следует ввести исправленное время удерживания 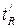
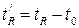 (1.4)
(1.4)
Величиной, не зависящей от скорости потока газа-носителя, является удерживаемый объем 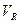 – это объем газа-носителя, который должен быть пропущен от момента ввода пробы до появления максимума пика на хроматограмме.
– это объем газа-носителя, который должен быть пропущен от момента ввода пробы до появления максимума пика на хроматограмме.
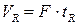 (1.5),
(1.5),
где 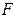 – объемная скорость потока, мл/с.
– объемная скорость потока, мл/с.
Объем для вымывания несорбируемого компонента 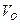 включает в себя объем колонки, не занятый сорбентом
включает в себя объем колонки, не занятый сорбентом
Приведенный удерживаемый объем 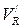 равен:
равен:
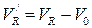 (1.6)
(1.6)
При постоянных условиях хроматографирования (скорость потока, давление, температура, состав фаз) значения характеристик удерживания строго воспроизводимы могут быть использованы для идентификации компонентов в качественном анализе и для физико-химических исследований.
Рассмотренные характеристики удерживания называются абсолютными. Кроме того, в хроматографии часто используют относительные характеристики удерживания.
При расчете относительного времени удерживания приведенное время удерживания какого-либо вещества относят к приведенному времени удерживания стандартного вещества:
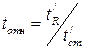 (1.7)
(1.7)
В качестве относительных параметров удерживания широко используют индексы Ковача. В отличие от относительных объемов индекс удерживания при выборе стандарта связан не с произвольно выбранным веществом, а с веществами, к которым предъявляют определенные требования. Во-первых, стандарт может представлять собой лишь нормальный алкан. Во-вторых, за стандарт берутся два соседних алкана, один из которых элюируется до, а другой после анализируемого соединения.
Индекс удерживания I какого-либо компонента рассчитывается по формуле:
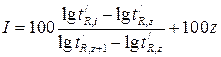 (1.8),
(1.8),
где 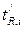 – приведенное время удерживания анализируемого i -го компонента;
– приведенное время удерживания анализируемого i -го компонента; 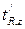 и
и 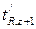 – приведенные времена удерживания н-алканов, z – число углеродных атомов.
– приведенные времена удерживания н-алканов, z – число углеродных атомов.
5. 5. Теоретические представления в газовой
хроматографии
В процессе хроматографического разделения часто происходит размывание пиков. Это явление обусловлено процессами, протекающими в колонке, например, медленностью сорбции и десорбции и др. Это приводит к тому, что разделение компонентов может вообще не произойти при значительной разнице в коэффициентах распределения.
Для объяснения специфического для хроматографии процесса размывания обычно используют теорию эквивалентных теоретических тарелок Мартина и Синджа и диффузионно-массообменную (кинетическую) теорию Ван-Деемтера.
5.5.1. Теория эквивалентных теоретических тарелок
По аналогии с теорией дистилляционных колонн хроматографическая колонка мысленно разбивается на ряд последовательных теоретических ступеней-тарелок, через которые периодически проходят порции газа. Каждая тарелка содержит подвижную (газовую) и неподвижную (жидкую или твердую) фазы. Предполагается, что за время нахождения порции газа на тарелке успевает установиться равновесие между подвижной и неподвижной фазами для всех компонентов. Таким образом, хроматографический процесс – многоступенчатый и состоит из большого числа актов сорбции и десорбции (в газо-адсорбционной хроматографии) или растворения и испарения (в газо-жидкостной хроматографии), а сама колонка рассматривается как система, состоящая из совокупности многих ступеней-тарелок.
Длина участка колонки, на которой достигается состояние равновесия между концентрацией вещества в подвижной и неподвижной фазах, называется условно высотой, эквивалентной теоретической тарелке (ВЭТТ). Существует простая зависимость:
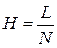 (1.9),
(1.9),
где L – длина хроматографической колонки, см; N – число теоретических тарелок; Н – высота, эквивалентная теоретической тарелке, см.
Для вычисления числа теоретических тарелок (ч.т.т.) измеряют ширину хроматографического пика на половине высоты, w 1/2. Тогда
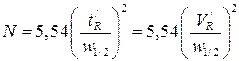 (1.10),
(1.10),
где w 1/2 –ширина полупика, выраженная в единицах времени (мин) или объема, мл.
Количественной мерой размывания хроматографических полос, т.е. эффективности колонки является ВЭТТ. С увеличением числа т.т. эффективность хроматографической колонки возрастает.
В теории теоретических тарелок реальный хроматографический процесс заменен идеальным, по которому хроматографическая полоса размывается вследствие равновесных процессов между подвижной и неподвижной фазами. Такое рассмотрение размывания хроматографической полосы не вскрывает сущности процесса и не дает информации о том, как подобрать такие условия, которые позволили бы уменьшить величину Н и тем самым повысить эффективность хроматографической колонки.
5.5.2. Кинетическая теория
В теории скоростей не делается допущений о наличии равновесных условий в колонке. Эта теория рассматривает динамику процесса распределения вещества между двумя фазами. Согласно упрощенному уравнению Ван-Деемтера, величина Н зависит от ряда параметров хроматографической колонки:
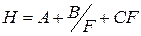 (1.11),
(1.11),
где Н – высота, эквивалентная теоретической тарелке, см; F – линейная скорость прохождения газа-носителя через колонку, см/с; А, В, С – константы.
Согласно этому уравнению, размывание хроматографической полосы, а следовательно, и снижение эффективности разделения связаныс тремя основными факторами:
1. Вихревая диффузия (слагаемое А). Хроматографическая колонка заполняется твердым зерненым сорбентом, поэтому газ-носитель перемещается по колонке через множество взаимосвязанных каналов. Одни молекулы могут продвигаться более короткими путями, другие – более длинными. Время пребывания последних молекул в колонке соответственно возрастает, что приводит к размыванию хроматографического пика. Вихревая диффузия уменьшается, если частицы твердого сорбента имеют одинаковые размеры и правильную форму и колонка плотно упакована.
2. Молекулярная диффузия. Слагаемое В/F характеризует размывание пиков, вызываемое диффузией анализируемого вещества в газе-носителе. (Диффузия в жидкой фазе ничтожно мала.) При бесконечно малой скорости газа-носителя вещество перемещается вниз по колонке под действием собственной молекулярной диффузии. Величина В/F, а следовательно, и Н в этом случае бесконечно большие.
3. Сопротивление массопередаче (член СF). Эта величина характеризует скорость распределения вещества между газом-носителем и неподвижной жидкой фазой. Чем меньше толщина слоя жидкости на твердом носителе и чем меньше вязкость этой жидкости, тем быстрее устанавливается равновесие.
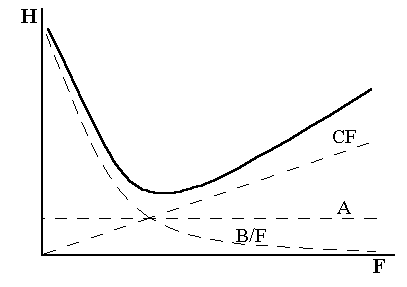 |
Рис.1.7. Графическое изображение уравнения Ван-Деемтера
Влияние каждого слагаемого уравнения (1.11) на величину Н в зависимости от линейной скорости подвижной фазы показано на рисунке 1.7.
При выборе колонки необходимо решать, уменьшить ли величину Н или уменьшить время разделения, повышая скорость потока газа-носителя, но сохраняя приемлемо малое значение Н. Член А не зависит от скорости газа-носителя и он весьма низкий в хорошо заполненной колонке. Член В/F с ростом скорости газа-носителя уменьшается, а СF, наоборот, увеличивается. Поэтому стараются подобрать такую скорость F, при которой В и С компенсировали бы друг друга, а величина Н была минимальной.
5.5.3. Оценка эффективност