 2015-02-14
2015-02-14 34069
34069Автор: преподаватель, Булатов М.И., Преподаватель, Санкт-Петербургский государственный технологический институт (технический университет)
1. Метод сравнения, основанный на сравнении аналитических сигналов (А) исследуемого и стандартного растворов.
Сущность метода. Приготавливают анализируемый и стандартный растворы аналита в светопоглощающей форме и измеряют их оптические плотности при одинаковых условиях:
Ax = elcxl и A ст= elc ст l
Разделив эти уравнения связи почленно одно на другое, получим:
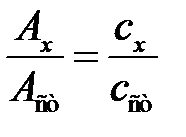 , откуда
, откуда 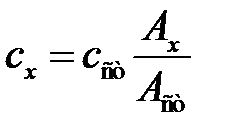 . (2.11)
. (2.11)
Метод применяется при однократных определениях и при неустойчивости аналитического сигнала во времени. Метод сравнения требует обязательного соблюдения основного закона светопоглощения, т.е. прямой пропорциональной зависимости между оптической плотностью раствора и его концентрацией.
2. Метод градуировочного графика
Сущность метода. Приготавливают 4-5 стандартных растворов аналита в светопоглощающей форме в интервале ожидаемых концентраций анализируемых растворов и измеряют их оптические плотности, не менее 3-х параллельных измерений для каждой концентрации. По средним значениям полученных данных строят градуировочный график зависимости А от с, с помощью которого и определяют неизвестную концентрацию аналита в анализируемом растворе. Метод не требует строгого соблюдения основного закона светопоглощения и применяется при многократных серийных анализах.
3. Метод добавок
Сущность метода. В мерной колбе приготавливают раствор аналита. Из одной его аликвотной части приготавливают анализируемый раствор с неизвестной концентрацией сх и измеряют его оптическую плотность Ах. К другой аликвотной части добавляют известное содержание определяемого элемента и в том же объеме готовят анализируемый раствор с добавкой. Затем при тех же условиях и в той же кювете измеряют оптическую плотность приготовленного раствора с добавкой Ах+д. Из отношений измеренных оптических плотностей получим:
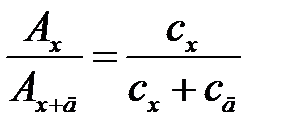 , откуда
, откуда
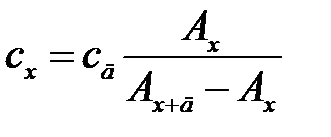 , (2.12)
, (2.12)
где сд – концентрация добавленного аналита в объеме фотометрируемого раствора.
Метод добавок применяют в тех случаях, когда наблюдается сильный матричный эффект (влияние основы пробы), который нельзя предусмотреть при приготовлении стандартных растворов. Являясь разновидностью метода сравнения, он требует обязательного соблюдения основного закона светопоглощения. Кроме того, чтобы не произошло «потери точности при вычитании в уравнении (2.12)», добавку аналита необходимо вводить такую, чтобы Ах+д ≈2 Ах. Можно использовать и графический варинт этого метода, но тогда необходимо приготовить два раствора с двумя разными добавками 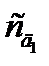 и
и 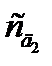 . По измеренным значениям оптической плотности Ах,
. По измеренным значениям оптической плотности Ах, 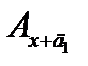 и
и 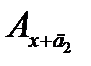 строят график (рис. 2.20). Модуль отрезка 0 сх и является неизвестной концентрацией сх.
строят график (рис. 2.20). Модуль отрезка 0 сх и является неизвестной концентрацией сх.
| с, мкг/мл |
| А |
| сх |
| Ах |
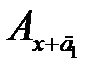 |
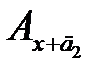 |
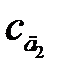 |
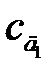 |
Рисунок 2.20 – Графическое определение концентрации аналита методом добавок: 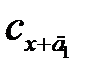 и
и 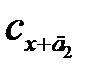 – концентрации анализируемого раствора с добавками д 1 и д 2, соответственно, а
– концентрации анализируемого раствора с добавками д 1 и д 2, соответственно, а 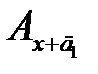 и
и 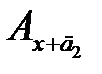 – соответствующие им значения оптических плотностей; 0 сх – графическое значение определяемой концентрации аналита сх
– соответствующие им значения оптических плотностей; 0 сх – графическое значение определяемой концентрации аналита сх
Сущность метода состоит в том, что оптические плотности анализируемых и стандартных растворов измеряют не по отношению к чистому растворителю с нулевым поглощением света, а по отношению к раствору определяемого элемента с известной концентрацией с 0, близкой к концентрации исследуемого раствора. Метод применяют при определении больших содержаний элементов, а так же для устранения мешающего влияния посторонних компонентов и исключения поглощения реагента. В этом методе измеренная относительная оптическая плотность А ¢ представляет собой разность оптических плотностей фотометрируемого раствора и раствора сравнения:
А ¢ х = Ах – А 0 = el (сх – с 0) l,
А ¢ст = А ст – А 0 = el (с ст – с 0) l.
Неизвестную концентрацию сх определяют либо как и в методе сравнения
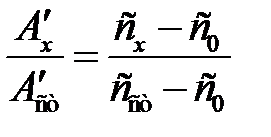 , откуда
, откуда
сх = с 0 + А ¢х(с ст – с 0)/ А ¢ст, (2.13)
либо используют градуировочный график, в начале координат которого концентрация с 0 (рис. 2.21).
| с |
| А´ |
| с 0 |
Рисунок 2.21 – Градуировочный график для определения концентрации растворов дифференциальным методом при прямом порядке измерений (сх, с ст> с 0); А´ – относительная оптическая плотность раствора; с 0 – концентрация нулевого раствора
Существуют и другие варианты этого метода, среди которых так же широко применяются два следующих.
В первом из них концентрация раствора сравнения (нулевого) больше концентрации анализируемых растворов (с 0 > с ст, сх). В этом варианте применяют обратный порядок измерений: анализируемые растворы условно принимают за раствор сравнения и по отношению к ним измеряют оптическую плотность раствора сравнения. Максимальная концентрация анализируемых растворов ограничивается концентрацией с 0 раствора сравнения. Концентрацию сх исследуемого раствора определяют аналогично предыдущему варианту с той лишь разницей, что здесь относительная оптическая плотность исследуемого раствора равна разности оптической плотности между значениями оптической плотности раствора сравнения и анализируемого раствора: А ¢ х = А 0– Ах. Концентрацию сх исследуемого раствора рассчитывают по формуле (2.14):
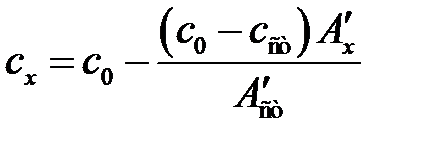 . (2.14)
. (2.14)
Если для определения неизвестной концентрации сх используют градуировочный график, то его строят в координатах А ¢ст= f (с 0– с ст) (рис. 2.22). При А ¢ х =0 сх = с 0 и при D с =0 сх = с 0, а при D сх = с 0 сх =0.
| с 0 |
| А'´ |
| Δ с = с 0– сх (ст) |
| с 0 |
| с 0(ст) |
Рисунок 2.22 – Градуировочный график для определения концентрации растворов дифференциальным методом при обратном порядке измерений (сх, с ст< с 0); А´ – относительная оптическая плотность раствора; Δ с = с 0– сх (ст) – разность концентраций аналита в нулевом и анализируемых растворах, соответственно
Сочетание первых двух вариантов реализуется в двустороннем или полном дифференцировании, где объединены прямой (сх ,ст> с 0) и обратный (с 0> сх ,ст) порядки дифференциальных измерений при фотометрическом определении веществ.
При фотометрировании анализируемых растворов, концентрации которых больше, чем концентрация раствора сравнения (сх ,ст> с 0), значения относительной оптической плотности берут, как обычно, со знаком плюс (+). Если концентрации анализируемых растворов меньше, чем концентрация раствора сравнения (сх ,ст< с 0), то полученные при фотометрировании растворов значения относительной оптической плотности берут со знаком минус (–).
Для построения градуировочного графика (рис. 2.23) приготавливают несколько стандартных растворов с концентрациями определяемого вещества большими, чем в растворе сравнения, и столько же стандартных растворов с концентрациями, меньшими, чем в растворе сравнения.
Измеренные относительные оптические плотности первой серии растворов откладывают на положительной полуоси графика, а второй серии – на отрицательной. Концентрацию исследуемого раствора определяют с помощью такого графика, используя положительную или отрицательную полуоси в зависимости от прямого или обратного порядка измерений относительной оптической плотности исследуемого раствора.

Рисунок 2.23 – Градуировочный график для определения концентрации раствора методом двухстороннего (полного) дифференцирования: А ´ – относительная оптичекая плотность раствора при сх (ст)> с 0 (со знаком плюс), а при сх (ст)< с 0 (со знаком минус)
Концентрации исследуемого раствора можно также рассчитывать и по формулам (2.13) и (2.14) при прямых и обратных дифференциальных измерениях оптической плотности раствора, соответственно.
Двустороннее дифференцирование при использовании одного и того же раствора сравнения позволяет увеличить интервал анализируемых концентраций примерно в 2 раза при неизменной воспроизводимости результатов.
5. Экстракционно-фотометрический метод
Экстракционно-фотометрический метод основан на сочетании экстракции светопоглощающей формы аналита не смешивающимся с водой органическим растворителем с последующим фотометрическим определением его в органической фазе одним из ранее описанных способов. Метод обладает высокой чувствительностью и селективностью.
Определяемый ион Ме+ с помощью экстракционного реагента НR в органическом растворителе, не смешивающимся с водой, экстрагируют в органическую фазу по реакции:
Ме+ + НR(о) + H2O? МеR(о) + Н3O+ (2.15)
Константу равновесия реакции экстракции
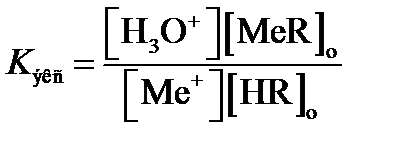 (2.16)
(2.16)
называют константой экстракции. Здесь [HR]o и [MeR]o – соответственно, равновесные концентрации экстракционного реагента и экстрагируемого соединения в органической фазе, моль×л–1; [Me+] – равновесная концентрация экстрагируемого аналита в водном растворе после завершения экстракции, моль×л–1.
Учитывая, что отношение [MeR]o/[Me+] характеризует коэффициент распределения D, из уравнения (2.16) получим приближенное уравнение, используемое при прогнозировании условий количественной однократной экстракции аналита по реакции (2.15):
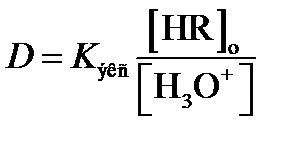 . (2.17)
. (2.17)
Количественное проведение однократной экстракции аналита достигается при условии, что коэффициент распределения D экстрагируемого соединения MeR удовлетворяет соотношению: D ≥102 W / V, где W и V – объемы водной и органической фаз, соответственно.
6. Фотометрическое титрование
Фотометрическое титрование выполняется аналогично титрованию с визуальным индикатором, но одновременно измеряется по мере титрования и оптическая плотность титруемого раствора. По результатам титрования строят кривую титрования (рис. 2.24) в координатах А = f (V R), где V R – добавляемый объем титранта R. Если при титровании образуется устойчивый комплекс (β MeR≥106), то на кривой титрования наблюдается резкий излом, по абсциссе которого определяют эквивалентный объем титранта V экв. При образовании малоустойчивого комплекса резкого излома на кривой не наблюдается и V экв определяют по абсциссе точки пересечения касательных к линейным участкам кривой. При этом значение V экв получается завышенным на величину? V.
Зная расход титранта и состав образующегося светопоглощающего соединения, находят массовое содержание аналита:
m Me = с R V экв M Me/ n, мг
где с R – молярная (формульная) концентрация реагента – титранта, моль/л; n – число эквивалентности – число молекул реагента в составе светопоглощающего соединения, связанных с одним ионом аналита Ме; V экв – эквивалентный объем реагента-титранта, мл; M –молярная масса определяемого аналита Ме.
| V R |
| А |
| 1.0 |
| 0.5 |
| 1.5 |
| 2.0 |
| Δ V |
Рисунок 2.24 – Кривая фотометрического титрования с образованием высокопрочного (1) и малоустойчивого (2) комплексов: Δ V – завышение эквивалентного объема титранта при образовании малоустойчивого комплекса
На рис. 2.24 показана кривая титрования, где свет поглощает только продукт реакции титрования MeR. Возможны и другие формы кривых титрования в зависимости от того, какой компонент фотометрической реакции обладает светопоглощением.
Метрологические характеристики
Метрологические характеристики в общем виде были рассмотрены в разделе 1.2.3. Они в полной мере применимы и к фотометрическим методам анализа, где в качестве аналитического сигнала Y следует использовать оптическую плотность А.
2.5 ИНФРАКРАСНАЯ СПЕКТРОСКОПИЯ
Инфракрасная спектроскопия (ИКС) принадлежит к обширной группе методов молекулярной спектрометрии и основана на избирательном поглощении излучения в инфракрасной области (0.8–1000 мкм) спектра. От других фотометрических методов она существенно отличается определенной особенностью в связи с тем, что способностью поглощать инфракрасное (ИК) излучение могут только те молекулы веществ и соединений, у которых изменяется дипольный момент при колебаниях атомов. Кроме того, поглощаемое ИК-излучение расходуется только на изменение колебательной и вращательной энергии молекулы, не вызывая из-за недостатка поглощаемой энергии (hν) электронных переходов.
ИК-спектры более сложные, чем электронные спектры в видимой области, поскольку б?льшая часть поглощенной энергии затрачивается именно на колебательные процессы. Поэтому ИК-спектры молекул характеризуются высокой информативностью и являются такой же характеристикой вещества, как отпечатки пальцев человека.
ИКС применяется в различных областях физико-химических исследований. Широкое применение ИКС получила в аналитической химии органических соединений. В неорганической химии она применяется, главным образом, при идентификации комплексных соединений, при установлении координационной связи атома металла с донорными атомами лигандов.
Происходжение инфракрасных спектров
Химические связи между атомами в молекуле не являются абсолютно жесткими и отдельные атомы находятся в постоянном движении, обусловливая колебание молекулы как единого целого. Все колебательные движения молекулы можно разложить на так называемые нормальные колебания, имеющие свои собственные пространственные характеристики и частоты. Нормальным называют такое молекулярное колебание, при котором все атомы движутся в одной фазе и с одинаковой частотой.
Различают два основных типа нормальных колебаний:
1. Валентные колебания – периодическое возвратно-поступательное движение атомов вдоль оси связи (симметричное и асимметричное), не изменяющее формы молекулы. Частота валентных колебаний атомов (атомных систем) в молекуле зависит от массы атомов и соединяющих их сил связи, ее можно рассчитать с помощью уравнения Гука, описывающего движение гармонических осцилляторов:
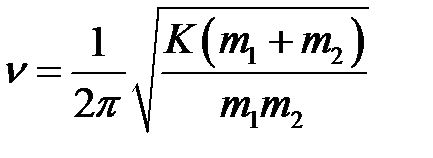 ,
,
где v – частота колебаний, с–1 (Гц); m 1 и m 2 – массы атомов, участвующих в образовании химической связи; К – коэффицент силы связи (силовая постоянная), дн×см–1.
2. Деформационные колебания, которые бывают, в основном, двух типов. Первый тип колебаний обусловлен изменением угла связи между двумя атомами, каждый из которых связан с третьим атомом. Второй тип – это движение группы атомов относительно остальных атомов молекулы.
Деформационные колебания бывают как симметричные, так и асимметричные, и многие из них направлены перпендикулярно линии связи. Среди других видов деформационных колебаний выделяют также «ножничные», «маятниковые», «веерные» и «крутильные». Схематическое изображение различных видов колебаний молекулы показано на рис. 2.25.
Для того, чтобы ИК-излучение могло взаимодействовать с колеблющейся молекулой, она должна претерпевать изменение дипольного момента в процессе колебания. Поглощается только такое ИК-излучение, с которым колеблющаяся молекула может когерентно взаимодействовать, т.е. частота которого совпадает с частотой колебания атомов в молекуле.
Рисунок 2.25 – Примеры различных типов колебаний молекулы:
а – валентные колебания изолированных связей и групп связей; б – деформационные колебания; 1 – симметричное; 2 – асимметричное; 3 – веерное; 4 – маятниковое; 5 – крутильное; 6 – ножничное. Знак (+) показывает движение за плоскость чертежа, а знак (–) – от плоскости чертежа вперед
Поглощенное ИК-излучение приводит к измененеию колебательной и вращательной энергии, а на осуществление электронных переходов ее недостаточно. Тем не менее одновременное изменение колебательной и вращательной энергий и межмолекулярные взаимодействия приводит к тому, что в ИК-спектре появляются не отдельные линии, а много полос поглощения, хотя и существенно более узких, чем в электронных спектрах (см. рис. 2.26). При этом интенсивность полосы поглощения тем больше, чем более полярная связь. Нормальные колебания обычно мало подвержены влиянию со стороны соседних частиц, так что ИК-спектр является в высшей степени характеристичным для данной молекулы.
В ИК-спектре выделяют три области, в каждой из которых можно получить различную информацию, используя различающуюся аппаратуру и технику эксперимента. Ближняя ИК-область (0.8–3 мкм) используется, в основном, для изучения колебаний атомов водорода в С–Н, N–H, O–H и подобных им связях. Для анализа органических соединений особенно часто используют среднюю ИК-область (2.5–15 мкм), т.к. здесь проявляется большое число полос поглощения функциональных групп. В дальней ИК-области (>15 мкм) имеют место низкочастотные колебания и вращения, полосы поглощения которых не находят широкого аналитического применения.
Рисунок 2.26 – Инфракрасный спектр пленки полистирола с линейной шкалой длин волн (а) и с линейной шкалой волновых чисел (б)
В ИК-спектре многоатомных молекул проявляются колебания двух типов: скелетные колебания, в которых атомы «скелета» молекулы участвуют примерно в одинаковой степени, и колебания характеристических групп, при которых сильные смещения испытывает лишь малая группа атомов молекулы.
Таблица 2.2 – Характеристические полосы поглощения некоторых атомных групп
| Группа | Приближенное значение волнового числа полосы поглощения, см–1 | Группа | Приближенное значение волнового числа полосы поглощения, см–1 |
| 1750–1600 | |||
| 1200–1000 | |||
| 2970 (асим. вален.) 2870 (сим. вален.) 1460 (асим. деформ.) 1375 (сим. деформ.) | |||
| 2930 (асим. вален.) 2860 (сим. вален.) 1470 (деформ.) | 800 – 600 | ||
| 600 – 500 | |||
| ~ 500 | |||
Полосы поглощения скелетных колебаний наблюдаются в интервале волновых чисел 1400–700 см–1 и характерны для линейных и разветвлено-цепных структур молекул. И хотя отдельные полосы поглощения очень трудно отнести к определенным колебаниям атомов, зато в целом совокупность наблюдаемых полос поглощения достаточно характеризует исследуемую молекулярную структуру. Частоты колебаний характеристических групп, напротив, мало зависят от строения молекулы в целом, и в основном, находятся в ближней ИК-области (0.8–3 мкм), не перекрывающейся с областью скелетных колебаний и могут быть использованы для аналитических целей. Именно по характеристическим частотам определенных атомных групп и проводят идентификацию органических соединений и обнаружение тех или иных атомных группировок и радикалов. В таблице 2.2 приведены характеристические полосы поглощения некоторых атомных групп и радикалов органических соединений.
Приведенные в таблице 2.2 полосы поглощения характерны для отдельных локализованных колебаний частей молекулы, частоты которых не слишком близки к частотам скелетных колебаний. Так, локализованные кратные связи (например, >C=C< или –CºC–) имеют частоты, которые являются весьма характерными. Однако когда две такие обособленные группы, имеющие сравнимые частоты, оказываются в молекуле рядом, возникает резонансное взаимодействие, значительно сдвигающее наблюдаемые частоты относительно ожидаемой величины. Так обособленная карбонильная группа в кетоне (R2C=O) и двойная связь >C=C< имеют характеристические полосы поглощения при 1715 и 1650 см–1, соответственно, а когда они находятся рядом, образуя структуру >C=C=C=O, их индивидуальные полосы смещаются соответственно к 1675 и 1600 см–1 и интенсивность полосы >C=C< увеличивается, становясь сравнимой с принципиально более интенсивной полосой >C=O. При более сильной связи этих групп, как, например, в радикале кетена >C=C=O, наблюдается поглощение вблизи 2100 и 1100 см–1, которое уже очень сильно сдвинуто относительно характеристических полос поглощения обособленных групп.
Сдвиги частот характеристических групп могут возникать и по другим причинам, в частности в результате взаимодействия между различными молекулами. Так, валентная частота группы –OH в спиртах сильно зависит от силы образующейся водородной связи, которая удлиняет и ослабляет связь –ОН и, следовательно, понижает ее частоту. Если водородная связь образуется между группой –ОН и, скажем, карбонильной группой, то частота карбонильной групп также понижается, хотя и в меньшей степени, чем у –ОН, поскольку водородная связь ослабляет связывание и в группе >C=O. Однако уже сами по себе сдвиги частот характеристических групп, обусловленные резонансными или межмолекулярными эффектами, являются высоко характеристическими и весьма полезны для аналитических целей.
Источники и приемники ИК-излучения
В качестве источников ИК-излучения используют не лампы накаливания, а специальные нагреватели – штифт Нернста, глобар и нихромовую ленту. Штифт Нернста изготавливают из оксидов циркония, иттрия и тория, он требует внешнего предварительного разогрева до 800–1000 °С. Его рабочая температура около 1500 °С. Глобар, изготовленный из карбида кремния, который имеет рабочую температуру 1200–1300 °С, напротив, требует постоянного охлаждения контактов. Нихромовая лента, состоящая из сплава никеля и хрома, обеспечивает рабочую температуру 1200–1500 °С. Приемником излучения, фиксирующим степень ослабления ИК-излучения, прошедшего через поглощающий слой, являются различные тепловые элементы: термопара, фотосопротивление, термосопротивление (болометр).
Измерение поглощения ИК-излучения
Для измерения поглощения ИК-излучения, как и в других методах молекулярной спектрометрии, используют два типа приборов (ИК-спектрометров) – однолучевые и двухлучевые. Однолучевой прибор фиксирует только один пучок ИК-излучения и непосредственно регистрирует его количество, прошедшее через анализируемый образец и оптику прибора (рис. 2.27). Пропуская ИК-излучение последовательно через кювету без образца и с анализируемым образцом, измеряют, соответственно, интенсивности ИК-излучения I 0 и I.
Количественный анализ Рисунок 2.27 – Принципиальная блок-схема однолучевого ИК-спектрометра: 1 – источник ИК-излучения; 2 –устройство выделения спектрального интервала (монохроматор); 3 – кюветное отделение; 4 – приемник излучения (фотосопротивление); 5 – электроника приемника; 6 – индикатор выходного сигнала. Во многих приборах монохроматор 2 помещают после кюветного отделения 3
В двухлучевых приборах ИК-излучение от источника разделяется на два одинаковых параллельных потока, один из которых проходит через анализируемый образец, а другой – через кювету сравнения (фон). Преобразованные световые потоки регистрируются детектором и регистрирующее устройство автоматически фиксирует их разность. Двухлучевые приборы удобны в работе, превосходят однолучевые в скорости регистрации ИК-спектров, но уступают им в точности полученных результатов. Однолучевой прибор позволяет получить более точные измерения пропускания, чем двухлучевой, и надежнее фиксировать любые его изменения. Поэтому он больше подходит для количественных определений.
В ИК-спектрометрах вся оптика, а также кюветы, в которых находятся растворы светопоглощающих соединений, должны быть изготовлены из «прозрачных» для ИК-излучения материалов, чаще всего из кристаллов галогенидов щелочных металлов (NaCl, KBr). В некоторых случаях, когда регистрируют поглощение ИК-излучения, примыкающего к видимой области, используют плавленый кварц и специальные стекла (см. табл. 2.3).
Определенные ограничения существуют и у применяемых растворителей. Например, верхний предел пропускания у хлороформа составляет 3.5 мкм, а у тетрахлорметана – 6.0 мкм. Вода, как растворитель, вообще не применяется, т.к. она в любых формах интенсивно поглощает ИК-излучение, и кроме того, несовместима с элементами спектрометров из кристаллов галогенидов щелочных металлов. При измерении поглощения ИК-излучения, кроме растворов, применяют твердое таблетирование с KBr и суспендирование с вазелином и другими маслами.
Таблица 2.3 – Верхние границы пропускания ИК-излучения (в мкм) некоторых материалов
| Материал | Длина волны, мкм | Материал | Длина волны, мкм |
| Стекло | ≤ 2.5 | CsBr | ≤ 15 |
| Плавленый кварц | ≤ 4.0 | CsI | ≤ 40 |
| NaCl | ≤ 16.0 | CaF2 | ≤ 10 |
| KBr | ≤ 25 | BaF2 | ≤ 13 |
| AgCl | ≤ 22 |
Количественное определение органических веществ по поглощению ИК-излучения проводят аналогично фотометрии в ультрафиолетовой и видимой областях. Аналитическим сигналом является оптическая плотность A =lg(I 0/ I)=–lg T, где Т – пропускание ИК-излучения анализируемым образцом (раствором). Здесь также при количественных определениях используется основной закон светопоглощения в его логарифмической форме: А= elcl и правило аддитивности оптических плотностей. Наиболее распространенным способом (методом) количественного определения является метод градуировочного графика.
Большое значение в количественной ИК-спектроскопии имеет неизбирательное поглощение фона, особенно при использовании твердых брикетированных образцов. Например, в модельном спектре (рис. 2.2) кроме характеристических полос 1-3, обусловленных поглощением ИК-излучения определенными группами, на всем протяжении спектра наблюдается неизбирательное поглощение фона – Δ. Для того, чтобы определить поглощение, обусловленное только анализируемым компонентом, из экспериментально измеренного поглощения необходимо исключить величину поглощения фона. В первом приближении эту задачу можно решить с использованием двухлучевых приборов и специальных фоновых стандартов, по отношению к которым снимаются ИК-спектры.
| Т, % |
| l |
| b |
| a |
| A |
| B |
| C |
| Δ |
Рисунок 2.28 – Определение величины пропускания вещества (без фона) методом базисных линий: ab – линия, соединяющая начало и конец полосы поглощения; Δ – поглощение фона
Для более точного исключения поглощения фона, особенно когда оно на разных участках спектра неодинаковое, применяют специальные приемы, самым простым из которых является так называемый метод базисных линий. В этом методе (см. рис. 2.28) соединяют прямой линией ab начало и конец полосы поглощения (1) и при длине волны l максимума поглощения определяют величину пропускания Т =ВС/АС и затем определяют оптическую плотность А =–lg Т. Затем по стандартным образцам строят градуировочный график А = f (с), с помощью которого и определяют неизвестное содержание аналита. Возникает вопрос, всегда ли необходимо пользоваться этим приемом? Как уже отмечалось, с небольшими погрешностями фоновое поглощение можно автоматически исключить применением двухлучевых спектрометров в сочетании с фоновыми стандартами. Если же ИК-спектры получают с помощью однолучевых спектрометров, то возможны следующие варианты.
1. Если анализируемый компонент имеет в спектре отдельную и достаточно интенсивную полосу поглощения аналита, например полосу 2 (рис. 2.28), а неизбирательное поглощение фона в стандартных и исследуемых образцах одинаковое, то нет необходимости отдельно определять поглощение фона. В этом случае градуировочный график смещается вверх параллельно на величину оптической плотности фона А ф (см. рис. 2.29) и на погрешности определения это практически не скажется.
| А |
| с |
| Ах+ф |
| Ах |
| Аф |
| сх |
Рисунок 2.29 – Градуировочный график для определения концентрации вещества с учетом (1) и без учета (2) неизбирательного поглощения фона (Аф)
2. Если при тех же условиях оптическая плотность аналита соизмерима (или даже меньше) с величиной плотности фона, то из-за возрастающих погрешностей целесообразно применять метод базисных линий.
3. При определении двух компонентов, когда их полосы поглощения в спектре накладываются, учет неизбирательного поглощения фона становится обязательным. Это требование в полной мере относится к фотометрированию и на двухлучевых приборах. В этом случае для дифференцированного определения каждого из аналитов используют метод Фирордта, решают систему линейных уравнений, выражающих суммарное поглощение обоими аналитами при двух длинах волн:
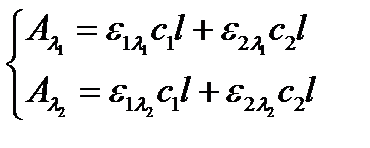 (2.18)
(2.18)
Согласно правилу аддитивности оптических плотностей, общая (суммарная) оптическая плотность анализируемой смеси поглощающих компонентов равна сумме парциальных оптических плотностей самих компонентов. В правой части системы уравнений (2.18) выражена сумма парциальных оптических плотностей компонентов, поэтому и экспериментально измеренная оптическая плотность смеси этих компонентов (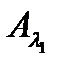 ,
, 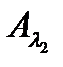 ) должна быть точно такой же, т.е. без оптической плотности фона.
) должна быть точно такой же, т.е. без оптической плотности фона.
2.6 ЛЮМИНЕСЦЕНТНЫЕ МЕТОДЫ АНАЛИЗА
2.6.1 Основные понятия
Люминесцентные методы анализа основаны на явлении люминесценции – свечении различных частиц, возбуждаемых каким-либо источником энергии. Такими частицами могут быть атомы, ионы, молекулы и более сложные соединения, которые при обратном спонтанном переходе из возбужденного состояния в нормальное излучают часть поглощенной энергии в виде квантов света. Традиционно к люминесцентным методам относят только молекулярную флуоресценцию в ультрафиолетовой и видимой областях спектра, а люминесценцию возбужденных атомов выделяют отдельно, как атомно-флуоресцентный анализ.
Академик С.И. Вавилов и В.Л. Левшин в процессе люминесценции различают свечение дискретных центров, когда люминесцирует та же самая частица, которая получила энергию при возбуждении, и рекомбинационные процессы, в которых излучение испускает другая частица, получившая энергию от возбужденной.
Люминесцентные методы чаще всего классифицируют по способу возбуждения и в этой связи различают:
- радиолюминесценцию (возбуждение радиоактивными частицами);
- рентгенолюминесценцию (возбуждение рентгеновскими лучами);
- электролюминесценцию(возбуждение электрическим полем);
- хемилюминесценцию (возбуждение экзотермической химической реакцией);
- фотолюминесценцию (возбуждение ультрафиолетовым или видимым светом).
В фотолюминесценции различают быструю спонтанную флуоресценцию, медленную вынужденную флуоресценцию и фосфорисценцию. Разновидности фотолюминесценции характеризуются различным «временем жизни» возбужденных состояний молекулы. Так, быстрая флуоресценция мгновенно прекращается с прекращением возбуждения молекулы, а фосфорисценция продолжается еще некоторое время и после прекращения возбуждения. Различие «времени жизни» возбужденных молекул при спонтанной флуоресценции (10–7 – 10–9 сек) и фосфорисценции (10–2 – 100 сек) объясняется различным механизмом перехода молекул из возбужденного состояния в основное.
Процессы, происходящие при возбуждении молекулы и люминесценции, наглядно можно иллюстрировать диаграммой Яблонского (рис. 2.30) При нормальных условиях невозбужденная молекула находится в основном колебательном состоянии v 0 на электронном уровне S 0 с минимальной энергией и возбуждение происходит именно с него. Два более высоких энергетических уровня S 1 и Т 1 соответствуют электронно-возбужденным синглетному и триплетному состояниям. Наиболее устойчивым энергетическим состоянием из них является синглетное, в котором спины (направление вращения электронов вокруг оси) электронов, занимающих одну и ту же орбиталь, антипараллельны ( ). Электронные переходы без изменения спина называют синглет-синглетными.
). Электронные переходы без изменения спина называют синглет-синглетными.
| КР |
| ВК |
| ИК |
| КР |
| ВК |
| ВК |
| ν 1 |
| ν 2 |
| s 1 |
| s 2 |
| ν 2 |
| ν 1 |
| ν 1 |
| ν 2 |
| s 0 |
| ν 2 |
| ν 1 |
| T 1 |
Рисунок 2.30 – Схема изменений энергетического состояния гипотетической молекулы при ее возбуждении (диаграмма Яблонского). На рисунке поглощение возбуждающего света показано прямыми стрелками, направленными вверх, а испускаемое люминесцентное излучение – стрелками вниз
Триплетное состояние является метастабильным, в котором спины электронов параллельны ( ).
).
Спонтанные синглет-синглетные переходы с возбужденного уровня S 1 на один из колебательных подуровней (v 0, v 1, v 2, …) основного состояния являются наиболее вероятными (в квантовомеханической терминологии «разрешенными»). В результате этого перехода испускается квант энергии hv, а сам процесс называют флуоресценцией.
В растворах избыточная энергия возбужденных молекул очень быстро (~10–13 с) теряется при столкновениях с окружающими молекулами. Этот процесс называется колебательной релаксацией (на рис. 2.30 он показан вертикальными волнистыми стрелками). Столь же быстро происходит переход молекулы с нижнего колебательного уровня верхнего возбужденного состояния на верхний колебательный уровень более низкого состояния. Этот процесс называют внутренней конверсией. Еще одним безызлучательным процессом дезактивации молекул является интеркомбинационный переход (на рис. 2.30 он показан горизонтальными волнистыми стрелками) молекулы с нижнего колебательного уровня верхнего возбужденного синглетного состояния S 1 на верхний колебательный уровень триплетного состояния Т 1. На этот метастабильный уровень молекула попадает, потеряв значительную часть энергии, полученной при возбуждении, и продолжает ее терять при колебательной релаксации, особенно при столкновениях с молекулами растворенного в воде кислорода. Поэтому вероятность перехода с триплетного уровня на основной очень мала (переход «запрещен»). Но тем не менее, такие переходы все-таки имеют место при фосфоресценции, при этом испускается квант света значительно меньшей энергии, чем при флуоресценции.
В некоторых случаях с участием Т 1-состояния может осуществляться еще один излучательный процесс – вынужденная замедленная флуоресценция, которая происходит в результате поступления к молекуле в Т 1-состоянии дополнительной энергии при столкновении частиц и при термической активации. При этом дополнительно возбужденная молекула переходит из Т 1-состояния в S 1-состояние с последующим излучением из него. Длительность свечения такой замедленной флуоресценции соизмерима с фосфоресценцией, а по энергии излучения она не отличается от спонтанной флуоресценции. Вынужденная флуоресценция характерна для сложных органических молекул и наблюдается при низких температурах или в очень вязких средах.
Небольшую часть люминесцентного излучения, обусловленную переходом возбужденной молекулы с самого первого возбужденного состояния S 1 на состояние S 0 без потери энергии называют резонансной люминесценцией (флуоресценцией).
Спектры люминесценции
Спектрами люминесценции называют графическое изображение распределения интенсивности люминесцентного излучения (I л) по длинам волн или частотам: I л= f (l) или I л= f (v). В отличие от атомно-флуоресцентных линейчатых спектров излучения спектры молекулярной люминесценции имеют форму широких полос излучения с полушириной полосы испускания ~ 50-100 нм, аналогично электронным спектрам поглощения, и графически изображается зависимостью I л= f (l).
Как уже отмечалось ранее, вклад энергии колебательных подуровней в общую энергию электронных уровней, как основного, так и возбужденного состояний молекулы приводит к появлению в спектрах люминесценции широких полос (50-100 нм) излучения (рис. 2.31). В то же время существуют оптимальные экспериментальные приемы, позволяющие значительно уменьшить ширину полос в спектрах люминесценции. Это прежде всего, регистрация спектров люминесценции при глубоком охлаждении до температуры жидкого азота (метод Шпольского). Но особенно эффективен метод синхронного сканирования спектров. Синхронные спектры – это спектры, получаемые при одновременном изменении длин волн возбуждения и испускания с постоянным сдвигом Δl между ними. С помощью современных спектрофлуориметров можно регистрировать синхронные спектры с шириной полосы излучения 8-10 нм. Узкий, резко выраженный пик полосы излучения в синхронном спектре получается только в той области, где перекрываются спектры возбуждения (поглощения) и излучения (люминесценции), и характерен для спектров с отчетливо выраженной колебательной составляющей энергии молекулы. Для получения оптимального результата поддерживается небольшая постоянная разность длин волн (Δl≈3-5 нм), хотя во многих случаях получаются узкие пики и при сдвиге Δl, равном разности между максимумами длин волн испускания и поглощения. Достоинство этого метода заключается в хорошем разрешении полос испускания смеси люминесцирующих соединений, что очень важно при идентификации органических веществ.
| I л |
| l, нм |
| el |
| П |
| l R |
| Л |
Рисунок 2.31 – Спектры поглощения (П) и люминесценции (Л) молекул: l R – длина волны резонансной люминесценции. Области оптимальных длин волн поглощения и люминесценции заштрихованы
Из схематического изображения процесса люминесценции (рис. 2.30) видно, что только переход между нижними колебательными подуровнями возбужденного и основного состояний (S 1→ S 0) имеет одну и ту же энергию поглощения и испускания, когда наблюдается резонансная люминесценция (lвозб=lлюм), а для остальных электронно-колебательных переходов энергия квантов поглощаемого излучения больше, чем энергия испускаемых (hv возб> hv люм). Поэтому максимумы в спектре люминесценции сдвинуты в область б?льших длин (меньших частот) по сравнению с положениями максимумов в спектрах поглощения возбуждающего излучения (закон Стокса и Ломмеля).
Расстояние Δl между максимумами в спектрах поглощения и люминесценции называют стоксовым смещением, которое у фосфоресценции больше, чем у флуоресценции. Чем больше стоксово смещение, тем легче отделить возбуждающий свет и устранить влияние фона на измерение люминесцентного излучения.
В редких случаях, когда возбуждаемая молекула до поглощения света обладала значительным запасом колебательной энергии, которая, суммируясь с энергией поглощенных квантов, может вызвать люминесценцию с меньшей длиной волны излучения, чем у возбуждающего света; при этом наблюдаются антистоксово свечение.
| I л |
| l |
| el |
Рисунок 2.32 – Зеркальная симметрия спектров поглощения и люминесценции раствора родамина 6Ж: 1 – спектр поглощения; 2 – спектр люминесценции
Важной характеристикой спектров возбуждающего и люминесцирующего излучений является их зеркальная симметрия (правило Левшина) – спектры поглощения и люминесценции зеркально симметричны относительно прямой, проходящей перпендикулярно оси длин волн (или частот) через точку их пересечения (см. рис. 2.32). Зеркальная симметрия характерна, в основном, для сложных молекул. Она позволяет по спектру люминесценции определить оптимальный интервал длин волн возбуждающего света.
Выход люминесценции
Способность к фотолюминесценции сложных молекул определяется, главным образом, их структурой. Наиболее важным фактором, обусловливающим возможность люминесценции молекул, является наличие у них жесткой и плоской структуры. Появлению жесткой компланарной структуры способствует образование в молекуле циклов или внутримолекулярных водородных и координационных связей. Тенденцию к образованию флуоресцирующих комплексов проявляют, как правило, катионы, не обладающие хромофорными свойствами (Al3+, Be2+, Ca2+, Mg2+, Zn2+ и др.). Напротив, катионы переходных металлов, обладающие хромофорными свойствами (Fe3+, Cu2+, Co2+, Ni2+, Cr3+ и др.) значительно реже образуют флуоресцирующие комплексы, так же в подобных комплексах преобладает безызлучательная дезактивация энергии возбуждения.
Интенсивность люминесценции. Эффективность преобразования энергии возбуждения (Е п) в энергию излучения (Е л) характеризуют абсолютным энергетическим (φ эн) и квантовым (φ кв) выходом люминесценции:
φ эн = Е л/ Е п и φ кв = N л/ N п,
где N л и N п – число излучаемых и поглощенных квантов света, соответственно.
Поскольку энергия кванта света E = hv (где h – постоянная Планка, а v – частота излучения), легко можно найти соотношение между энергетическим и квантовым выходом люминесценции:
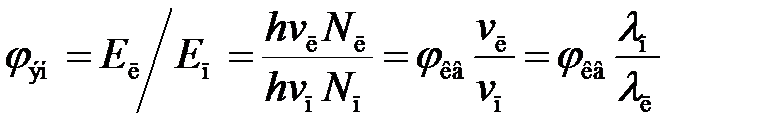 . (2.19)
. (2.19)
Из уравнения (2.19) видно, что величина φ эн зависит от длины волны поглощаемого при возбуждении света. Согласно закону Вавилова, при возбуждении флуоресценции излучением коротковолновой части спектра величина энергетического выхода φ эн до некоторого предела (до начала перекрывания спектров поглощения и люминесценции) увеличивается пропорционально длине волны возбуждающего излучения, а квантовый выход φ кв в этом интервале длин волн постоянен. Интенсивность флуоресценции зависит от количества поглощенной при возбуждении световой энергии и квантового выхода. Чем больше квантовый выход, тем больше интенсивность флуоресценции при одинаковом количестве поглощенной энергии Е п. Однако на практике приходится иметь дело с относительным квантовым выходом люминесценции, который пропорционален абсолютному квантовому выходу и оценивается по измеряемой величине интенсивности люминесценции. Относительный квантовый выход всегда меньше абсолютного (теоретического) из-за неизбежных потерь энергии излучения как вследствие проявления различных видов тушения люминесценции, так и при ее экспериментальной регистрации.
При низких концентрациях люминесцирующего вещества интенсивность люминесценции I л пропорциональна числу излучаемых квантов N л:
I л = k л N л = k л φ кв N п,
где k л – коэффициент пропорциональности.
Число поглощенных при возбуждении квантов N п пропорционально интенсивности поглощенного возбуждающего света, которая определяется по разности интенсивности до его поглощения (I 0) и прошедшего через поглощающий слой (I). Согласно основному закону светопоглощения интенсивность прошедшего через поглощающий слой света равна:
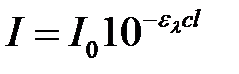 . (2.20)
. (2.20)
Соответственно,
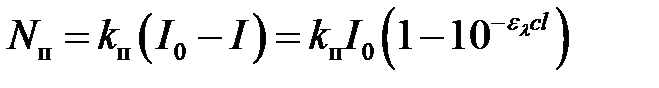 .
.
Следовательно,
 . (2.21)
. (2.21)
В результате разложения степенной функции 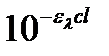 в ряд Тейлора получаем:
в ряд Тейлора получаем:
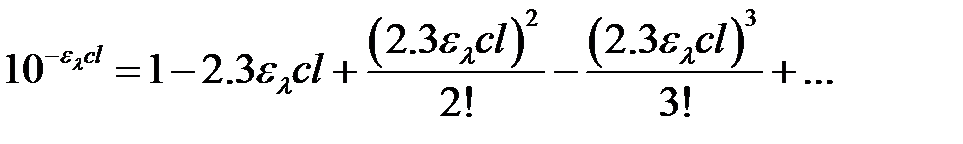 . (2.22)
. (2.22)
В типичном для проявления люминесценции случае минимальных концентраций люминесцирующего вещества произведение elcl <<10–2. Поэтому в уравнении (2.22) третьим и последующими слагаемыми можно пренебречь и уравнение (2.21) преобразовать:
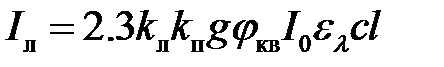 , (2.23)
, (2.23)
где el – молярный коэффициент поглощения возбуждающего света, л×моль×см–1; l – толщина поглощающего слоя, см; с – молярная концентрация люминесцирующего вещества; g – приборный фактор (коэффициент), показывающий какая часть люминесцирующего излучения регистрируется прибором; k л, k п – коэффициенты пропорциональности (k л k п<1).
Объединив все постоянные величины в уравнении (2.23) в одну, в окончательном виде получим:
I л = kc. (2.24)
Линейная зависимость I л от с лежит в основе всех прямых количественных люминесцентных определений аналитов.
Тушение (гашение) люминесценции
Под тушением люминесценции понимают любой процесс физической или химической природы, который приводит к уменьшению интенсивности люминесценции. К их числу относятся все процессы, сопровождающиеся безызлучательными потерями молекулой избыточной энергии, полученной ею при возбуждении. Природа таких процессов различная. Это и внутреннее тушение вследствие внутримолекулярных взаимодействий, и внешнее, которое может происходить при межмолекулярном взаимодействии с молекулами посторонних веществ, присутствующих в растворе. Известны также температурное тушение (при повышении температуры), концентрационное тушение (при значительном увеличении концентрации люминесцирующего вещества) и тушение растворителем.
В наиболее общем случае внешнего тушения примесями выделяют, в основном, два вида, условно называемых статическим (I рода) и динамическим (II рода). При статическом тушении постороннее вещество взаимодействует с невозбужденными молекулами активируемого соединения (люминофора), образуя новые химические соединения, которые либо вообще не люминесцируют, либо люминесцируют в другой спектральной области и с другим квантовым выходом.
При динамическом тушении постороннее вещество взаимодействует с возбужденными молекулами с образованием продуктов, не способных к люминесценции, что неизбежно приводит к уменьшению числа центров свечения и интенсивности люминесценции.
Иногда имеет место так называемый эффект внутреннего фильтра – уменьшение относительного выхода люминесценции, обусловленное частичным поглощением возбуждающего или испускаемого излучения (или того и другого одновременно) молекулами растворенных веществ и растворителя.
Важное для аналитической практики значение имеет концентрационное тушение (или самотушение) люминесцирующего вещества. Линейная зависимость I л= f (c) соблюдается только при очень малых концентрациях люминесцирующего вещества (в среднем 10–5–10–8 моль×л–1) и при увеличении концентрации линейная зависимость нарушается вследствие самопоглощения излучения раствором люминесцирующего вещества, а при достижении пороговой концентрации (~10–4 моль×л–1) при дальнейшем ее увеличении интенсивность люминесценции даже может уменьшиться (см. рис. 2.33). Причины наблюдаемого концентрационного эффекта могут быть как физическими (реабсорбция люминесцирующего излучения), так и химическими, например, образование нелюминесцирующих ассоциатов.
| I л |
| с |
| с п |
Рисунок 2.33 – Зависимость интенсивности люминесценции I л от концентрации с люминесцирующего вещества; с п – пороговая концентрация
Измерение интенсивности люминесценции
Принципиальная схема измерения интенсивности люминесценции показана на рис. 2.34.
Рисунок 2.34 – Принципиальная блок-схема флуориметра: 1 – источник возбуждающего света; 2, 4 – светофильтры; 3 – кювета с анализируемым раствором; 5 – фотоэлемент или фотоумножитель; 6 – усилитель фототока; 7 – регистрирующее устройство
Излучение от источника излучения (1) направляется на светофильтр или монохроматор (2) к кювете с анализируемым раствором (3). Возникающее люминесцентное излучение проходит через светофильтр (или монохроматор) (4) и попадает на фотоэлемент или фотоумножитель (5), в котором излучение преобразуется в фототок и после его усиления усилителем (6) регистрируется измерительным прибором (7). Назначение светофильтров (или монохроматоров) такое же, как и в фотометрическом анализе: светофильтр 2 выделяет (пропускает) свет такого интервала длин волн, который максимально поглощается возбуждаемыми молекулами, а светофильтр 4 – выделяет наиболее интенсивную спектральную область флуоресцирующего излучения. Оба светофильтра предназначены обеспечивать максимальную чувствительность флуориметрических определений. Часто для уменьшения погрешностей, связанных с нестабильностью интенсивности возбуждающего света, измеряют не абсолютную интенсивность флуоресценции, а относительную – по отношению к какому-либо внутреннему стандарту с максимумом флуоресценции в другой области. В некоторых случаях в качестве внутреннего стандарта используют точно зафиксированную часть возбуждающего излучения (опорный сигнал) в сочетании с модулятором.
2.6.2 Количественное флуориметрическое определение веществ
Главная область применения молекулярного флуоресцентного анализа – это высокочувствительные количественные определения в интервале концентраций 10–7–10–9 моль×л–1. Аналитическим сигналом при количественных определениях является интенсивность флуоресценции (люминесценции) I фл. Теоретическая чувствительность, вытекающая из уравнения (2.23), характеризуется минимальной определяемой концентрацией с мин:
с мин = I фл(мин)/2.3 gI 0 φ кв εll, (2.25)
где I фл(мин) – минимальное значение интенсивности флуоресценции, которое может быть надежно зарегистрировано используемым измерительным прибором (флуориметром, спектрофлуориметром).
Как видно из уравнения (2.25), для повышения чувствительности желательно использовать более интенсивные источники возбуждения (I 0) и в той спектральной области, где возбуждающее излучение максимально поглощается активируемыми молекулами (εl – максимальный), и стрем

