 2015-04-01
2015-04-01 1212
1212Верапамил и дилтиазем блокируют кальциевый ток в тканях с медленными потенциалами действия: синусовом и АВ-узле и приводят к сдвигу в положительную сторону порога возбуждения, что обуславливает:
- замедление деполяризации в АВ-узле, что приводит к увеличению ЭРП, замедлению проведения, на ЭКГ вызывает удлинение интервала PQ. Клинически это приводит подавлению ритма по типу re-entry, включающий АВ узел, а также снижению проведения предсердных экстрасистол к желудочкам.
- замедление спонтанной диастолической деполяризации, что проявляется угнетением автоматизма пейсмейкерных клеток СА-узла, а на ЭКГ – снижением ЧСС, хотя выраженность этого эффекта выражена существенно меньше, чем при применении БАБ;
Так же, как под воздействием БАБ, снижение входа кальция в клетку в фазу II ПД не оказывает существенного влияния на длительность ПД. В то же время уменьшение поступления кальция в клетку приводит к снижению сократимости миокарда, поэтому их назначение не показано при застойной сердечной недостаточности.
Фармакокинетика. Верапамил и дилтиазем являются липофильными препаратами, почти полностью всасываются, но значительно метаболизируются при первом прохождении через печень. Поэтому биодоступность составляет верапамила 20-35%, а дилтиазема – 45%, а при в/в введении эффект верапамила будет выше по сравнению с преоральным применением той же дозы препарата. При повторном применении верапамила и дилтиазема биодоступность может повысится, элиминация замедлится. Верапамил и дилтиазем метаболизируется в печени. Их метаболиты обладают меньшей терапевтической активностью. Период полувыведения дилтиазема составляет от 2 до 7 ч; при однократном приеме верапамила - 2,8-7,4 ч, при его длительном применении – 4,5-12 ч. В плазме верапамил связывается с альбумином и α1-кислым протеином, а дилтиазем кроме того - с γ-глобулином. Связь с белками плазмы верапамила составляет более 90%, дилтиазема – 80-85%. Метаболиты верапамила в основном выводится почками, дилтиазема – через желудочно-кишечный тракт. При нарушенной функции печени биодоступность и период полувыведения верапамила увеличиваются. У дилтиазема наблюдается двойной пик концентрации препарата в крови, что связано с кишечно-печеночной рециркуляцией препарата.
Циклоспорин может замедлять всасывание верапамила и дилтиазема. При одновременном их назначении с сердечными гликозидами верапамил и дилтиазем могут вытеснять из связи с белками плазмы дигоксин, усиливая его действие, что может привести к гликозидной интоксикации. Учитывая то, что препараты метаболизируются с участием изофермента 3А4 цитохрома Р450, при назначении с кетоконазолом, флуконазолом, эритромицином, грейпфрутовым соком, которые его ингибируют, возможно усиление гипотензивного действия. При применении с теофиллином возможно повышение концентрации теофиллина, и как следствие – симптомов его передозировки. При назначении с 600мг рифампицина, фенобарбиталом являющихся индукторами изофермента 3А4, эффект перорально назначенного препарата практически нивелируется.
Верапамил и дилтиазем усиливают гипотензивное действие других гипотензивных препаратов. При применении с БАБ возможно нарушение АВ-проводимости. При применении с амиодароном возможна опасность остановки сердца. При применении с препаратами Iа группы усиливается отрицательный хронотропный, инотропный и дромотропный эффект, что может приводить к развитие тахикардии вследствие антеградного проведения. При применении трициклических антидепрессантов возможно удлинение интервала QT, миорелаксантов – повышение их эффекта.
Показания: стенокардия напряжения, вазоспастическая стенокардия, артериальная гипертензия, наджелудочковые нарушения ритма: наджелудочковая тахикардия, мерцательная аритмия и трепетание предсердий;
Побочные эффекты: желудочковая тахикардия при WPW-синдроме брадикардия, т.к. верапамил и дилтиазем тормозя АВ-проведение, они усиливает предвозбуждение по дополнительным проводящим путям; артериальная гипотония, особенно при в/в введении, усиление признаков сердечной недостаточности, могут возникать сердцебиения, синоатриальная блокада, нарушения атриовентрикулярной проводимости, асистолия, проаритмический эффект. артериальная гипотония, головная боль, головокружения, покраснение кожных покровов, сердечная недостаточность, пастозность в области лодыжек и голеней (тибиальные отеки), запоры, тошнота, рвота, необструктивная паралитическая кишечная непроходимость (очень редко), обратимые нарушения функции печени, аллергические реакции, потливость, мышечные судороги, переходящая спутанность сознания (редко), слабость, усталость, гиперплазия десен, гинекомастия, болезненные позывы на мочеиспускание, ухудшение функции почек; фоточувствительность, кожная сыпь – для дилтиазема.
Противопоказания: гиперчувствительность, беременность и кормление грудью, выраженный аортальный стеноз, брадикардия, слабость синусового узла, синоатриальная блокада, атриовентрикулярная блокада II-III степени без установленного электрокардиостимулятора, сердечная недостаточность, комбинация с БАБ у пациентов со значительно нарушенной функции левого желудочка.
Препараты III класса – блокаторы калиевых каналов.
За счет замедления выхода калия из клетки удлиняют реполяризацию, и соответственно рефрактерный период (рисунок 7). Это является основой их антиаритмического действия. На ЭКГ замедление реполяризации проявляется удлинением интервала QT. Так как реполяризация присуща всем клеткам – могут оказывать антиаритмическое действие при НРС любой локализации.
Основные представители этого класса – амиодарон и соталол - обладают также достаточно сильным бета-адреноблокирующим действием. Кроме того, амиодарону присуща также способность блокировать быстрые натриевые канала. Интересно, что блокада исходящего тока калия может ускорять МДД – поэтому влияние на ЧСС у препаратов этого класса зависит от баланса между их бета-адреноблокирующим действием (потенциально замедляющим ЧСС) и влиянием на калиевые каналы (потенциально увеличивающим ЧСС).
Рисунок 8. Влияние препаратов 3 класса на ПД в клетках сократительного миокарда
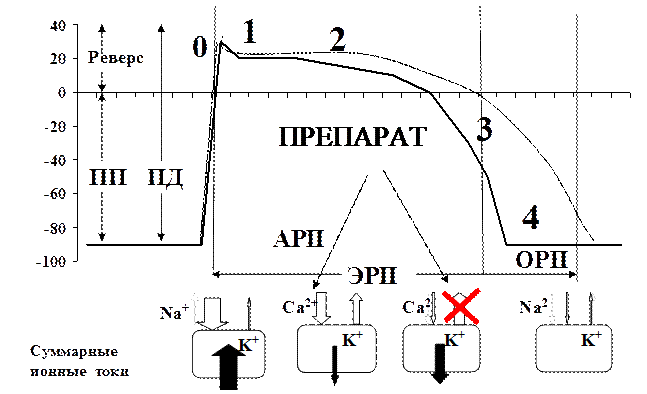
Рисунок 9. Влияние препаратов 3 класса на ПД в пейсмекерных клетках
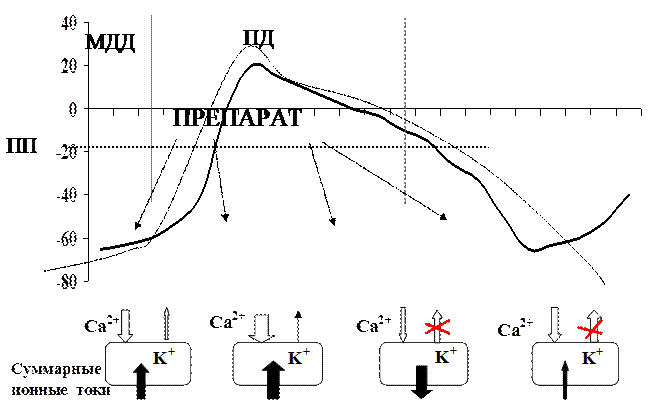
Амиодарон является структурным аналогом тиреоидных гормонов.
Фармакокинетика. Биодоступность амиодарона составляет 20-80%. Он высоко жирорастворим, поэтому медленно выводится и накапливается во многих тканях. Связь с белками плазмы составляет 96%. Он распределяется в жировой ткани, а также в печени, легких, селезенке. Учитывая то, что амиодарон большой кажущийся объем распределения, и его накопление в тканях, а следовательно, и достижение терапевтической концентрации происходит медленно в начале лечения используют высокие насыщающие дозы от 800 до 1600 мг/сут с последующий переходом на поддерживающую дозу. Т1/2 составляет от 2 до 10 дней, может увеличиваться при регулярном приеме и составлять до от 25 до 110 дней после его отмены. Метаболизируется в печени изоферментом цитохрома IIIA4, образующийся при этом метаболит дезэтиламиодарон обладает активностью, близкой к амиодарону. Амиодарон практически не выводится почками. При сопутствующих заболеваниях печени, почек и сердечной недостаточности дозу амиодарона не снижают. Амиодарон ингибирует изоферменты цитохрома IIIA4 и IIC9, поэтому при одновременном приеме флекаинида, прокаинамида, хинидина, варфарина и дигоксина необходимо снижать дозы этих препаратов.
Фармакодинамика. Учитывая механизм действия амиодарона, он обладает следующими эффектами:
- основной эффект – удлинение продолжительности ПД и рефрактерного периода во всей ткани миокарда, в том числе в дополнительных путях проведения, что связано с блокадой калиевых каналов;
- в небольшой степени тормозит быстры входящий натриевый ток, подобно лидокаину блокирует как открытые, так и инактивированные натриевые каналы. В отличие от препаратов I класса не вызывает значительного расширения интервала QRS;
- блокирует кальциевые каналы – свойство препаратов IV класса, в результате чего может появляться брадикардия и замедление проводимости в АВ-узле;
- обладает эффектом неконкурентного блокатора β- и α-адренорецепторов;
- вызывает дилатацию периферических сосудов, происходит снижение периферического сосудистого сопротивления и постнагрузки;
- дилатацию коронарных артерий, увеличивая коронарный кровоток
- может вызывать небольшой отрицательный инотропный эффект
- может изменять действие тироксина на сердце
Побочные эффекты. При приеме амиодарона часто возникает удлинение интервала QT, брадикардия, поражение щитовидной железы (которое может сопровождаться как гипотиреозом, так и тиреотоксикозом), появление микроотложений в роговице глаза, поражение легких. Наиболее тяжелые частые побочные реакции связаны с пневмосклерозом, фактором риска развития которого являются сопутствующие заболевания дыхательной системы и доза 400мг/сут и более. Для контроля возникновения пневмосклероза необходимо проводить исследование функции внешнего дыхания и рентгенографию. Также возможно нарушение функции печени, поражение нервов и мышц. При в/в введении может возникнуть гипотония, которая, вероятно, связана с вазодилатацией, снижением сердечного выброса, а также действием растворителя.
Противопоказания: слабость синусового узла, АВ-блокада II-III степени, фракция выброса менее 40 %, что связано с высоким риском внезапной смерти, гипокалиемия, особенно при сочетании с удлинением интервала QT – риск возникновения аритмий, в т.ч. типа torsades de pointes, гиперчувствительность, беременность, кормление грудью.
Соталол является препаратом III класса со свойствами β-адреноблокатора. Существуют два изомера соталола d- и l-соталол. Непосредственно β-адреноблокирующий эффект составляет 30-50% от эффекта пропанолола. При этом этим эффектом обладает l-изомер. Оба изомера вызывают увеличение продолжительности ПД, что регистрируется на ЭКГ как удлинение интервала QT, и ЭРП.
В настоящее время применяется d-соталол, так рацемическая смесь d-,l-соталола увеличивает риск смерти у больных после ИМ с фракцией выброса 31-40%.
Фармакокинетика. Соталол – неселективный водорастворимый β-адреноблокатор. У соталола биодоступность составляет 100%. При одновременном приеме с молочными продуктами биодоступность снижается. Соталол находится в свободном состоянии в плазме. Период полувыведения составляет 7-15 ч. Препарат не метаболизируется в печени. Выводится почками, следовательно кумулируется в организме. Начало антиаритмического действия отмечается через 1 час, максимальный эффект – через 2,5-4 часа, продолжительность эффекта 24 часа. При в/в введении начало действия – через 5 мин, длительность эффекта 90-120 мин.
Побочные эффекты соталола связаны с β-адреноблокирующей активностью (см. БАБ). Чаще возникают одышка, усталость, брадикардия, бронхоспазм, усиление признаков сердечной недостаточности, синдром отмены. С блокадой калиевых каналов связано возможное удлинения интервала QT, развитие аритмий типа torsades de pointes, часто возникающие после удлинения интервала QT, желудочковая тахикардия, чаще возникающая в первые 3 дня приема, особенно при дозе препарата более 640 мг и составляет 3,8% случаев, а также могут возникать лейкопения, эозинофилия.
Противопоказания. Блокады сердца II-III степени, синусовая брадикардия, кардиогенный шок, синдром удлинения интервала QT, наличие бронхоспазмов в анамнезе, обструктивные заболевания легких, бронхиальная астма, ацидоз, аллергические реакции в анамнезе, в т.ч. на соталол и сульфаниламидные препараты.
Дофетилид. Это высокоизбирательный блокатор калиевого тока задержанного выпрямления (Ikr), при котом ионы калия выходят из клетки, причем в фазу плато (фаза 2) амплитуда этого тока нарастает медленно, а фазу реполяризации (фаза 3) – быстро. Эффектом дофетилида является удлинение ПД и интервала QTc.
Фармакокинетика. При приеме внутрь биодоступность составляет более 90%.максимальная концентрация достигается через 2 ч. Период полувыведения составляет 7-13 ч. Метаболизируется в печени, но не взаимодействует с дигоксином, пропронололом, варфарином. Выводится почками. При тяжелой почечной недостаточности уменьшают дозу препарата, не назначается совместно с препаратами, усиливающими транспорт катионов.
Применяется для поддержания синусового ритма при мерцательной аритмии. В исследовании DIAMOND, в котором исследовалось влияние дофетилида на смертность и аритмии, показано, что дофетилид не влияет на смертность при тяжелой сердечной недостаточности и после инфаркта миокарда. Показана эго эффективность при восстановлении синусового ритма при мерцательной аритмии и трепетании предсердий.
Побочные эффекты. Учитывая, что дофетилид является высокоизбиротельным препаратом, он не вызывает внесердечных побочных реакций. Дофетилид вызывает пируэтную тахикардию в 1-3% случаев в первые 3 дня применения препарата, в связи с чем необходимо длительное мониторирование ЭКГ в этот период применения препарата.
Ибутилид - блокатор калиевого тока задержанного выпрямления (Ikr). В некоторых экспериментальных данных указывается, что он усиливает медленный натриевый ток эти механизмы приводят к удлинению ПД. Ибутилид выпускается в форме для в/в введения. Он быстро распределяется в большом объеме, незначительно связывается с белками плазы. Период полувыведения 2-12 ч. Подвергается значительному метаболизму при первом прохождении через печень, выводится почками. не взаимодействует с БАБ, БКК, варфарином, диуретиками.
Ибутилид применяют для быстрого восстановления синусового ритма при мерцательной аритмии и трепетании предсердий в первые несколько дней от начала пароксизма.
Побочным эффектом ибутилида является возникновение удлинения интервала QT, проаритмическое действие., проявляющийся пируэтной тахикардией.
Суммируя вышесказанное, можно вкратце перечислить механизм действия антиаритмических препаратов
• Класс I - блокаторы быстрых натриевых каналов - тормозят Na ток, замедляют скорость фазы 0 и амплитуду ПД → замедляют проводимость в предсердиях и желудочках, иногда расширяют QRS
• Класс IV - блокаторы медленных кальциевых каналов – блокируют вход Са в клетку - уменьшают амплитуду и укорачивают ПД клеток с медленным электрическим ответом, но удлиняют ЭРП за счет замедления восстановления функции канала
• Класс II - бета-адреноблокаторы – блокируют вход Са в клетку, угнетают фосфоламбан - уменьшают амплитуду ПД, замедляют скорость спонтанной диастолической деполяризации СУ и АВУ, удлиняют ЭРП АВУ, не влияют на ЭРП клеток сократительного миокарда, слегка укорачивают QT
• Класс III - блокаторы калиевых каналов - удлиняют реполяризацию → удлиняют ПД, ЭРП, QT, замедляют ЧСС, ускоряют МДД – повышают ЧСС
• СГ – блокаторы Na-K-АТФазы → способствует накоплению Na → замедляет выход Са из клетки → усиливает сокращения, снижает содержание К в клетке → укорачивает ПД; дигоксин - М-холиностимулятор → замедляет ЧСС

