 2018-01-21
2018-01-21 7251
7251
Сильные электролиты полностью диссоциируют на ионы. В водных растворах сильных электролитов существуют простые или сольватированные катионы и анионы. Это подтверждается современными физическими и физико-химическими исследованиями. Однако опытным путем найдено, что электропроводность водных растворов сильных электролитов не эквивалентна той электропроводности, которую можно было бы ожидать при 100%-ной диссоциации молекул на ионы. Такое несоответствие объясняется теорией сильных электролитов, предложенной Дебаем и Хюккелем. Согласно этой теории, в растворах сильных электролитов между ионами существует электростатическое взаимодействие. Каждый ион окружен ионной атмосферой из противоионов. Ионная атмосфера тормозит движение ионов в электрическом поле, в результате чего создается эффект неполной диссоциации сильного электролита.
Мерой электростатического взаимодействия ионов является ионная сила раствора μ, равная полусумме произведений квадрата заряда каждого из присутствующих в растворе ионов на его концентрацию.
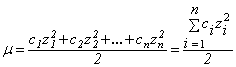 | (1) |
В связи с влиянием ионной силы раствора та концентрация, согласно которой ион действует в растворе, называется активной концентрацией (активностью):
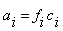 , , | (2) |
где f – коэффициент активности, который характеризует взаимодействие ионов.
Коэффициент активности может быть рассчитан в зависимости от ионной силы по выражениям:
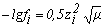 при µ <10-2 при µ <10-2 | (3) |
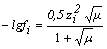 при 10-2 < µ < 10-1 при 10-2 < µ < 10-1 | (4) |
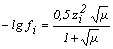 + 0, 1z2µ при 0,1 < µ < 1 + 0, 1z2µ при 0,1 < µ < 1 | (5) |
или взят из справочной литературы (необходимо объяснить учащимся, как пользоваться таблицей «Коэффициенты активности ионов при различной ионной силе»).
Активность необходимо использовать при расчете любого химического равновесия, если в системе находится сильный электролит, концентрация которого больше 1.10-4 моль/л.
РАСТВОРЫ ЭЛЕКТРОЛИТОВ, содержат в заметных концентрациях ионы-катионы и анионы, образующиеся в результатеэлектролитической диссоциации молекул растворенного в-ва. Р-ритель (чистый или смешанный) обычно в сколько-нибудь значит. степени не диссоциирован. Растворы электролитов обладают способностью проводить электрич. ток и относятся к проводникам второго рода. Благодаря увеличению общего числа частиц коллигативные св-ва бесконечно разбавленных растворов электролитов (т. е. св-ва, зависящие только от концентрации растворенного в-ва, но не от его природы) существенно отличаются от тех же св-в растворов неэлектролитов. Этим, в частности, объясняется увеличение осмотич. давления в сравнении со значением, предсказываемым законом Вант-Гоффа (см. Осмос), понижение давления пара р-рителя над р-ром в сравнении с предсказываемым Рауля законом и др. Наличиемионов обусловлены также классификация растворов электролитов, особенности теоретич. подходов в сравнении с др. классами р-ров. Наиб. изучены водные растворы электролитов, играющие важную роль во многих биол., геол. и техн. процессах. Неводные растворыэлектролитов служат средой для проведения синтеза и электрохим. процессов, используются в совр. технологиях (создание новыххимических источников тока, солнечных батарей, процессы разделения в-в и др.).
Классификация растворов электролитов основана на классификации электролитов. Соответственно о растворах электролитовговорят как о симметричных и несимметричных в зависимости от того, распадается ли молекула растворенного в-ва в р-ре на два ионаили на большее число частиц; z, z-зарядных [напр., р-р NaCl 1,1-зарядный, р-р СаС12 2,1-зарядный] и т.п.-По степени диссоциацииэлектролита , к-рая равна отношению числа молекул, диссоциированных на ионы, к полному числу молекул в р-рс, различают сильные ( = 1), слабые ( << 1) электролиты и, соотв., р-ры сильных и слабых электролитов. Такое деление, однако, является условным и отражает состояние электролита в р-ре, определяемое не только природой растворенного в-ва и р-рителя, но и концентрацией(молярной долей хэл), т-рой Т, давлением р.
В зависимости от состояния растворенного в-ва до растворения выделяют два класса растворов электролитов-р-ры ионофоров и р-ры ионогенов. Ионофоры в чистом состоянии существуют в виде ионных кристаллов (напр., галогениды щелочных металлов). В сильнополярных р-рителях (напр., в воде) ионофоры, как правило, диссоциируют полностью и составляют р-ры сильных электролитов. В слабополярных р-рителях (напр., в уксусной к-те) они образуют р-ры слабых электролитов. Ионогены до растворения состоят измолекул, они являются потенц. электролитами, электролитич. диссоциация проходит, как правило, в две стадии и обычно не полностью (хлорная к-та в уксусной к-те).
Растворители для растворов электролитов -как правило, полярные жидкости (чистые или смешанные). Чем больше диэлектрич. проницаемость р-рителя, тем значительнее ослабляется сильное электростатич. притяжение противоположно заряженных ионов, что способствует возникновению в р-ре ионов. Интенсивное взаимод. последних с молекулами р-рителя приводит к связыванию ионов смолекулами р-рителя (см. Сольватация). Важна также способность молекул р-рителя выступать в качестве доноров или акцепторовпротонов или электронов. В зависимости от этих двух св-в различают четыре группы р-рителей: 1) протонные р-рители (вода, спирты, карбоновыс к-ты и др.), к-рые являются хорошими донорами протона и обладают высокой диэлектрич. проницаемостью ( > 15); 2) апротонные дшюлярные р-рители (нек-рые апротонные амиды, кстоны, сульфоксиды и др.), обладающие высокой диэлектрич. проницаемостью, но не обладающие донорно-акцепторными св-вами в отношении протона; 3) электронодонорные-р-рители (напр., эфиры); 4) неполяр-ныс р-рители (сероуглерод, углеводороды), к-рые обладают низкой диэлектрич. проницаемостью ( < 15) и не обладают донорно-акцепторными св-вами ни по отношению к протону, ни по отношению к электрону.
Находящиеся в растворах электролитов ионы могут существовать в виде своб. сольватир. ионов или в виде ассоциатов-контактных или сольватно разделенных ионных пар, тройников и т.д. Поскольку ионные пары не проводят электрич. ток, содержание своб. ионов врастворах электролитов определяется по его электрич. проводимости, в то время как общее число ионов (свободных и ассоциированных) можно определить, напр., спектрофотомет-рич. методами.
Термодинамика растворов электролитов. Равновесные термодинамич. св-ва растворов электролитов описываются парциальными молярными величинами, в к-рых различают катионные и анионные вклады. Напр., для электролита типа 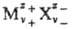 , состоящего из z+-валентных катионов Mz+ и z_-валентных анионов X-, хим. потенциал равен:
, состоящего из z+-валентных катионов Mz+ и z_-валентных анионов X-, хим. потенциал равен:
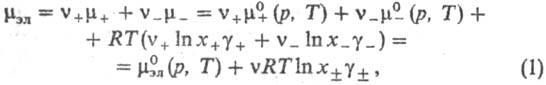
где v=v++v_, x+ и х--молярные доли катионов и анионов соотв., средняя молярная доля электролита хb = = 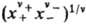 , + и _ -коэффициенты активности катионов и анионов,
, + и _ -коэффициенты активности катионов и анионов, 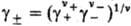 -средний коэф. активности электролита (см. Активность),
-средний коэф. активности электролита (см. Активность), 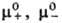 и
и 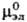 -стандартные хим. потенциалы катионов, анионов и электролита соотв., R-газовая постоянная.
-стандартные хим. потенциалы катионов, анионов и электролита соотв., R-газовая постоянная.
Др. парциальные молярные величины связаны с эл термо-динамич. соотношениями: парциальная молярная энтропия Sэл = -(9эл/9Т)p, парциальная молярная энтальпия Hэд_= = -Т2[(9эл/Т)/9Т]р, теплоемкость Ср = -Т(92эл/9Т2)p, парциальный молярный объем Vэл = (9эл/9р)T.
В качестве стандартного состояния компонентов в растворах электролитов выбирают: для р-рителя-состояние чистой жидкости, для растворенного в-ва-состояние в гипотетич. р-ре, где его концентрация и активность равны единице, а термодинамич. св-ва 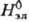 ,
, 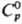 ,
, 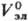 равны соответствующим значениям для бесконечно разб. р-ра.
равны соответствующим значениям для бесконечно разб. р-ра.
В электролитах с неполной степенью диссоциации (а < 1) выражение (1) заменяется соотношением:
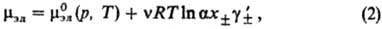
где 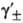 -средний ионный коэф. активности. Степень диссоциации а находят из условия хим. равновесия, к-рое в частном случае симметричного электролита (v+ =v_ = 1) приводит к ур-нию для константы диссоциации KD (или константы ассоциации Ка):
-средний ионный коэф. активности. Степень диссоциации а находят из условия хим. равновесия, к-рое в частном случае симметричного электролита (v+ =v_ = 1) приводит к ур-нию для константы диссоциации KD (или константы ассоциации Ка):
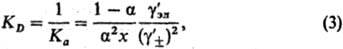
где 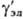 - коэф. активности недиссоциир. молекул (для разбавленных растворов электролитов
- коэф. активности недиссоциир. молекул (для разбавленных растворов электролитов 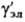 = 1). Аналогично (1) вводится также коэф. активности для р-рителя S, к-рый связан с ионными коэф. активности Гиббса- Дюгема уравнением.
= 1). Аналогично (1) вводится также коэф. активности для р-рителя S, к-рый связан с ионными коэф. активности Гиббса- Дюгема уравнением.
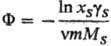
Для описания отклонения от идеального состояния разбавленных растворов электролитов используют кажущиеся осмотические коэффициенты, характеризующие отклонение осмотич. давления от значения, определяемого законом Вант-Гоффа, и связанные с ионными коэф. активности соотношением:
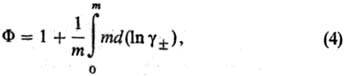
где m - моляльность электролита, b-ионные коэф. активности в моляльной шкале, MS-молярная масса (кг·моль-1), xS-молярная доля р-рителя.
Хим. потенциалы эл, коэф. активности b и осмотич. коэффициенты Ф м. б. определены экспериментально прямыми или косвенными методами: по давлению пара растворенного в-ва или р-рителя, по р-римости, по измерениям эдс электролитич. цепи. Из калориметрич. экспериментов находят парциальную молярную энтальпию Hэл, а из измерений плотности-парциальные молярные объемы Vэл. Поскольку измеримы только суммарные термодинамич. характеристики электролита, для катионов и анионов хим. потенциалы + и _, их стандартные значения 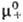 и
и 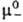 , коэф. активности + и _ и связанные с ними парциальные молярные величины м. б. определены только приближенно, на основе нетер-модинамич. допущений (напр., о равенстве вкладов одинаковых по размерам и степеням окислений катионов и анионов), путем экстраполяции эксперим. данных для различных растворов электролитов с общим катионом илианионом и др.
, коэф. активности + и _ и связанные с ними парциальные молярные величины м. б. определены только приближенно, на основе нетер-модинамич. допущений (напр., о равенстве вкладов одинаковых по размерам и степеням окислений катионов и анионов), путем экстраполяции эксперим. данных для различных растворов электролитов с общим катионом илианионом и др.
Наряду с хим. потенциалами ионов используют также электрохим. потенциал i-го иона с валентностью zi:

где F-Фарадея постоянная, -внутр. электрич. потенциал раствора электролита (см. Межфазные скачки потенциала).
Статистические теории растворов электролитов основаны на методах стати-стич. механики, их осн. задача-расчет св-в исходя из энергии межчастичного взаимодействия. Развиваются след. подходы: ионный подход (уровень Макмиллана-Майера); ион-но-молекулярный подход (уровень Борна-Оппенгеймера); электрон-ядерный подход (уровень Шрёдингера). Ионный подход является традиционным и к настоящему времени наиб. развит. Он основан на рассмотрении в явном виде только ионов, р-ритель из явного рассмотрения исключается, что требует усреднения ф-ции распределения Гиббса по всем мол. конфигурациям р-рителя (см.Статистическая термодинамика). Энергия межионного взаимод. представляется как сумма слагаемых унарного, бинарного, тернарного и т.д. типов. Унарные слагаемые выражаются через своб. энергию сольватации Wi i-го иона, характеризуемую изменением энергии Гиббса системы при переносе иона из идеальной газовой фазы в бесконечно разб. р-р. Значение Wi совпадает с неидеальной частью стандартного хим. потенциала i-го иона, причем выделяют электростатич., неэлект-ростатич. и хим. вклады в значение Wi. Электростатич. вклад Wiэл м. б. рассчитан согласно модели Борна, в рамках к-рой р-ритель рассматривается как бесструктурная среда, характеризуемая диэлектрич. проницаемостью :
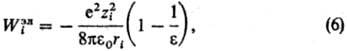
где 0-диэлектрич. постоянная (диэлектрич. проницаемость вакуума), ri-ионный радиус (см. Атомные радиусы). Для уточнения расчета вместо радиуса ri используется эффективный радиус 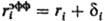 , где через i учитываются размеры молекул р-рителя. Предпринимаются попытки моделирования р-рителя путем введения вместо диэлектрич. ф-ции и учета нелинейных диэлектрич. эффектов.
, где через i учитываются размеры молекул р-рителя. Предпринимаются попытки моделирования р-рителя путем введения вместо диэлектрич. ф-ции и учета нелинейных диэлектрич. эффектов.
К неэлектростатич. вкладам в Wi относят индукционный и дисперсионный вклады (см. Дисперсионное взаимодействие), а также вклад, связанный с работой, к-рую необходимо затратить для образования в р-рителе полости и внедрения в нее иона. Расчет этих вкладов производится теми же методами, что и для р-ров неэлектролитов. Для расчета энергии сольватации применяют квантовохим. методы.
Бинарные слагаемые в энергии межионного взаимод. выражаются через потенциалы Wij(R), описывающие эффективное взаимод. ионовсортов i и j, находящихся на расстоянии R друг от друга. Соотв. тернарные слагаемые выражаются через потенциалы, описывающие трехчастичное взаимод. ионов и т. д. Учет бинарных и высших слагаемых в выражении для энергии межионного взаимод. позволяет описывать концентрац. зависимости термодинамич. св-в растворов электролитов.
Св-ва растворов электролитов характеризуются сложными концентрац. зависимостями, обусловленными конкуренцией вкладов разл. типов межчастичных взаимодействий. Обычно ограничиваются учетом потенциалов парного взаимодействия. Его существ. особенность-кулоновский характер межионного взаимод. на больших расстояниях (при 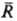 :,):
:,):
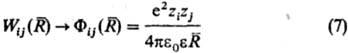
С кулоновским взаимод. связано экранирование межионных взаимод. и образование ионных комплексов, эти процессы характеризуются соотв. радиусом Дебая rD = 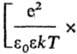
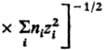 и радиусом Бьеррума
и радиусом Бьеррума 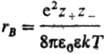 (k-пoстоянная Больцмана, ni-плотностьионов i-го сорта). Первый из них (rD) описывает расстояния, на к-рых экранируется поле иона за счет образования облака ионовпротивоположного знака, второй (rB)-расстояния, на к-рых кулоновское притяжение между катионом и анионом превышает среднюю энергию теплового движения, что приводит к образованию ионных пар. Эффекты экранирования кулоновских взаимод. учитываются Дебая - Хюккеля теорией; в этой теории первое приближение определяет ионные коэф. активности в области предельных разбавлений (ni: 0). Согласно теории Дебая - Хюккеля, коэф. активности ионов уменьшаются с концентрацией раствора электролита. Наличие врастворах электролитов ионных комплексов учитывается на основе представления о хим. равновесии между свободными и ассоциированными ионами, что приводит к ур-нию, аналогичному (3).
(k-пoстоянная Больцмана, ni-плотностьионов i-го сорта). Первый из них (rD) описывает расстояния, на к-рых экранируется поле иона за счет образования облака ионовпротивоположного знака, второй (rB)-расстояния, на к-рых кулоновское притяжение между катионом и анионом превышает среднюю энергию теплового движения, что приводит к образованию ионных пар. Эффекты экранирования кулоновских взаимод. учитываются Дебая - Хюккеля теорией; в этой теории первое приближение определяет ионные коэф. активности в области предельных разбавлений (ni: 0). Согласно теории Дебая - Хюккеля, коэф. активности ионов уменьшаются с концентрацией раствора электролита. Наличие врастворах электролитов ионных комплексов учитывается на основе представления о хим. равновесии между свободными и ассоциированными ионами, что приводит к ур-нию, аналогичному (3).
С увеличением концентрации электролита возникает необходимость учитывать и некулоновскую часть межионного взаимод., для чего прибегают к нек-рым моделям. При этом наряду с индукционным, дисперсионным, обменным и др. видами межчастичных взаимод. некулоновский потенциал учитывает сольватац. эффекты, связанные с влиянием р-ри-теля. В частности, учет некулоновской части взаимод. стабилизирует уменьшение коэф. активности ионов с концентрацией и может объяснить их увеличение, наблюдаемое экспериментально. Наипростейшей ионной моделью растворы электролитов является модель заряженных твердых сфер (т. наз. примитивная модель). Первые попытки описания примитивной модели были выполнены в рамках теории Дебая-Хюккеля (второе приближение). Более корректно учет размера ионов и неку-лоновского взаимод. осуществляется на основе методов статистич.термодинамики (см. Жидкость).
В рамках примитивной модели размеры ионов отличаются от кристаллографич. радиусов из-за сольватац. эффектов. Однако даже при одном и том же выборе размеров ионов удовлетворительно описать эксперим. результаты для разл. термодинамич. св-в растворахэлектролитов в примитивной модели оказалось затруднительным. К более корректным результатам приводит модель парного взаимод. типа "прямоугольной ямы", в к-рой ширина потенц. ямы выбирается равной диаметру молекулы р-рителя, а глубина ямы считается подгоночным параметром, учитывающим сольватац. эффекты; при этом используются кристаллографич. размеры ионов. В более реалистич. модели Фридмана в некулонов-ском потенциале межионного взаимод. выделяют три слагаемых, соответствующих: 1) главному отталкиванию ионов, определяемому их кристаллографич. размерами; 2) эффекту поляризации полости ионов р-рителем; 3) потенциалу Гер-ни-Франка, описывающему структурные эффекты, связанные с перекрыванием сольватных оболочек ионов при их сближении. Расчеты на основе ион-молекулярных моделей показывают, что на малых расстояниях межионные потенциалы имеют отталкивательный характер, на больших расстояниях, в соответствии с (7), зависят от расстояния между ионами асимптотически, как и при кулоновском взаимод., на промежут. расстояниях осциллируют вблизи этой асимптоты, причем с уменьшением размера иона (или с увеличением его валентности) амплитуда осцилляции возрастает, что соответствует усилению роли сольватац. эффектов.
Пренебрежение трехчастичными (и высшими) межионными взаимод. ограничивает возможности ионного подхода. В частности, для 1,1-зарядных водных растворов электролитов ионный подход обеспечивает количеств. описание термодинамич. св-в в областиконцентраций до 1 М. Учет концентрац. зависимости диэлектрич. проницаемости позволяет немного расширить эту концентрац. область. Формально расширения области применимости ионного подхода можно достигнуть, дополняя полученные с его помощью результаты разл. эмпирич. поправками. Примером такого подхода может служить полуэмпирич. ур-ние Питцера для осмотич. коэффициента или метод Робинсона-Стокса описания ионных коэф. активности с учетом гидратации (с помощью гидра-тац. чисел). Для описания многокомпонентных растворов электролитов широко используется правило Здановского, основанное на предположении о том, что смешение изописстич. р-ров разл. электролитов, химически не взаимодействующих между собой, происходит без измененияактивности р-рителя.
Ионно-молекулярный подход основан на рассмотрении в явном виде как ионов, так и молекул р-рителя. Главные результаты получены в 70-80-х гг. 20 в. на базе расчетных методов, интенсивно развиваемых в теории жидкостей. Это в осн. метод интегральных ур-ний для корреляц. ф-ций, метод кластерных разложений, теория возмущений, а также компьютерное моделирование. Благодаря явному учету ионно-молекулярных и межмолекулярных взаимод. возможно описание не только термодинамич., но и структурных св-в растворовэлектролитов. В частности, важный результат - описание сольватации ионов в зависимости от концентрации и др. параметров р-ра, объяснение концентрационных, температурных и барич. зависимостей св-в в широких интервалах состава, т-ры и давления.
Наипростейшей ион-молекулярной моделью растворов электролитов является ион-дипольная модель, в к-рой ионы рассматриваются как заряженные твердые сферы, а молекулы р-рителя моделируются твердыми сферами с дипольным моментом. Полученные выражения для термодинамич. ф-ций обобщают ур-ния, используемые в ионном подходе. В частности, в предельном случае малыхконцентраций выражения для ионных коэф. активности включают члены, основанные на теории Дебая-Хюккеля, а выражения для энергии сольватации-борновскую ф-лу (6) с эффективным радиусом иона 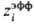 , в к-ром поправка i в явном виде зависит от диэлектрич. проницаемости р-рителя и соотношения размеров иона и молекулы. Выражение для диэлектрич. проницаемости удовлетворительно описывает эффект ее уменьшения при увеличении концентрации ионов.
, в к-ром поправка i в явном виде зависит от диэлектрич. проницаемости р-рителя и соотношения размеров иона и молекулы. Выражение для диэлектрич. проницаемости удовлетворительно описывает эффект ее уменьшения при увеличении концентрации ионов.
Предпринимаются попытки учета квадрупольного элект-рич. момента и поляризуемости молекул р-рителя, а также взаимод., ответственных за образование в растворах электролитов ассоциатов и сольватов. Наиб. реальные модели разработаны в осн. для водных растворов электролитов и базируются обычно на компьютерном моделировании. Для описания ионно-молекулярных и межмолекулярных взаимод. применяют эмпирич. модели воды (модель ST2, модель центр. сил и др.), а также модели, основанные на квантовохим. расчетах. Рассчитанные парциальные радиальные ф-ции распределения дают информацию о структуре р-ра. В частности, с помощью ионно-молекулярных ф-ций определяют координац. числа сольватации. Найденные с помощью парциальных радиальных ф-ций структурные факторы удовлетворительно согласуются с данными дифракц. измерений.
Электрон-ядерный подход основан на учете электроста-тич. взаимод. между электронами и ядрами, входящими в состав ионов имолекул в растворах электролитов. Этот подход является наиб. последовательным, он основан на квантовомех. рассмотрении и разработан пока лишь для ион-молекулярных комплексов.
Важное значение в физ. химии растворов электролитов имеют исследования транспортных св-в, особенно электрич. проводимости (см.Электропроводность электролитов). Наличие ионов заметно сказывается на диффузии, вязкости, теплопроводности.
Ионная сила раствора — мера интенсивности электрического поля, создаваемого ионами в растворе. Полусумма произведений из концентрации всех ионов в растворе на квадрат их заряда. Формула впервые была выведена Льюисом:
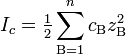 ,
,
где c B — молярные концентрации отдельных ионов (моль/л), z B заряды ионов
Суммирование проводится по всем типам ионов, присутствующих в растворе. Если в растворе присутствуют два или несколько электролитов, то вычисляется общая суммарная ионная сила раствора.
Например, для раствора NaCl с концентрацией 0,001 моль/л, в котором присутствуют два вида однозарядных ионов Na+ и Cl− с концентрациями также равными 0,001 моль/л, ионная сила будет вычисляться следующим образом:
I(NaCl) = 0,5(z²(Na+)•c(Na+) + z²(Cl−)•c(Cl−)) = 0,5(1²•c(NaCl) + (-1)²•c(NaCl)) = c(NaCl)
И ионная сила соответственно будет равна концентрации раствора:
I = 0.5(1²•0,001 моль/л + (-1)²•0,001 моль/л) = 0.5(0,001 моль/л + 0,001 моль/л) = 0,001 моль/л
Это верно для раствора любого сильного электролита, состоящего из однозарядных ионов. Для электролитов, в которых присутствуют многозарядные ионы, ионная сила обычно превышает молярность раствора.
Ионная сила раствора имеет большое значение в теории сильных электролитов Дебая — Хюккеля. Основное уравнение этой теории (предельный закон Дебая — Хюккеля) показывает связь между коэффициентом активности иона ze и ионной силы раствора I в виде:
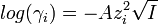 ,
,
где γ — коэффициент активности, А — постоянная, не зависящая от заряда иона и ионной силы раствора, но зависящая от диэлектрической постоянной растворителя и температуры.
Активность компонентов раствора — эффективная (кажущаяся) концентрация компонентов с учетом различных взаимодействий между ними в растворе, то есть с учетом отклонения поведения системы от модели идеального раствора.
| Содержание [убрать] · 1 Выбор стандартного состояния · 2 Методы определения активности o 2.1 По равновесному давлению пара o 2.2 По повышению температуры кипения раствора o 2.3 По понижению температуры замерзания раствора o 2.4 По осмотическому давлению раствора o 2.5 По распределению компонента между конденсированными фазами o 2.6 По равновесию химической реакции с газовой фазой o 2.7 По значению э. д. с. гальванического элемента · 3 Литература · 4 Ссылки |
Активность была предложена в 1907 году Льюисом как новая переменная, применение которой вместо концентрации позволяет использовать для описания свойств реальных растворов относительно простые уравнения, полученные для идеальных систем. Альтернативой этому пути является использование более сложных уравнений, учитывающих взаимодействие между частицами (см., например, уравнение Ван-дер-Ваальса).
Активность отличается от общей концентрации на некоторую величину. Отношение активности (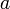 ) к общей концентрации вещества в растворе называется коэффициентом активности:
) к общей концентрации вещества в растворе называется коэффициентом активности:
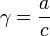
Коэффициент активности служит мерой отклонения поведения раствора (или компонента раствора) от идеального. Отклонения от идеальности могут быть обусловлены различными химическими и физическими причинами — дипольные взаимодействия, поляризация, образование водородных связей, ассоциация, диссоциация, сольватация и др.[1]
Исходя из понятия химического потенциала, активность компонента в растворе можно определить как величину, которую нужно подставить в выражения для химического потенциала компонента в идеальном растворе:
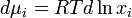
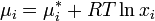
(где 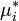 — химический потенциал чистого
— химический потенциал чистого 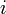 -го компонента) вместо мольной доли x, для того, чтобы получить действительное значение химического потенциала -го компонента в реальном растворе:
-го компонента) вместо мольной доли x, для того, чтобы получить действительное значение химического потенциала -го компонента в реальном растворе:
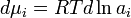
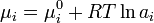
где 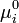 — стандартный химический потенциал.[2]
— стандартный химический потенциал.[2]
Размерность и величина активности зависит от используемого способа выражения концентрации — если 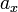 (активность при выражении концентрации как мольной доли) величина безразмерная, то
(активность при выражении концентрации как мольной доли) величина безразмерная, то 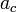 и
и 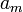 (для молярности и моляльности соответственно) — размерные величины, выражаются в моль/л и моль/кг.
(для молярности и моляльности соответственно) — размерные величины, выражаются в моль/л и моль/кг.
Коэффициент активности в общем случае может быть как больше, так и меньше единицы (при этом говорят о положительных или отрицательных отклонениях от идеального поведения соответственно, или о положительных и отрицательных отклонениях от закона Рауля). Возможны и знакопеременные отклонения от идеального поведения (то есть коэффициент активности меньше единицы при одних концентрациях, и больше — при других). Так, например, для железа в системе Fe-S при 1300 °C в[3] рекомендуются коэффициенты активности от 0,004 при 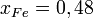 до 1,47 при
до 1,47 при 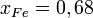 .
.
Отметим, что величина активности и коэффициента активности может быть различной в зависимости от выбора стандартного состояния.
[править]Выбор стандартного состояния
При использовании активности и коэффициента активности важную роль играет выбор стандартного состояния компонента, то есть состояния, в котором 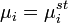
Для растворов взаимно неограниченно растворимых жидкостей в качестве стандартного может быть выбрано состояние чистого компонента (как для растворителя, так и для растворенного вещества):
при 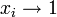
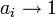 и
и 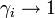
Иногда такой выбор называют симметричной системой стандартного состояния.
В случае, если рассматривается раствор газа и или твердого вещества в жидкости, мольную долю растворенного вещества нельзя изменять до единицы. Тогда для растворителя — жидкости — стандартное состояние может быть выбрано так же, как показано выше, а для растворенного вещества за стандартное состояние принимают гипотетический раствор с концентрацией, равной единице, но сохраняющий свойства предельно разбавленного раствора. Иначе говоря, это такое состояние, для которого давление пара численно равно константе Генри:
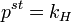
Таким образом, для растворителя и растворенного вещества здесь принимаются разные стандартные состояния — это несимметричная система стандартных состояний.
В системах с ограниченной растворимостью за стандартное может быть принято состояние компонента в насыщенном растворе:
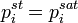
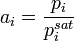
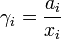
В засимости от исследуемой системы, в качестве стандартного может быть выбрано и другое состояние, например, для серы при исследовании богатых сульфидных расплавов — состояние серы в стехиометрическом сульфиде[3]. При рассмотрении результатов эксперимента, использовании справочных данных и т. п. следует обязательно указывать, какое именно состояние компонента принято за стандартное.
[править]Методы определения активности
Экспериментальные методы определения активности компонентов в растворе основаны на изучении какого-либо гетерогенного равновесия в системе. При рассмотрении этих методов следует помнить, что в условиях равновесия химические потенциалы i-го компонента в разных фазах (I и II) равны:
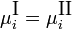
Это соотношение является исходной точкой для вывода расчетных уравнений в некоторых из методов определения активности. Кроме того, активности компонентов в некоторой фазе связаны между собой уравнением:
[править]По равновесному давлению пара
В основе этого метода лежит соотношение:
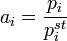
где 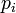 — парциальное давление пара компонента над раствором, а
— парциальное давление пара компонента над раствором, а 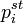 — давление пара этого компонента для стандартного состояния (см. выше). Соответственно, если за стандартное состояние принято состояние чистого компонента, то
— давление пара этого компонента для стандартного состояния (см. выше). Соответственно, если за стандартное состояние принято состояние чистого компонента, то 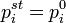 .
.
Эксериментальные методы определения давления пара компонентов над раствором весьма разнообразны; выбор того или иного из них определяется, в частности, исследуемой системой (водный раствор или иная низкотемпературная система, либо расплавленный металл, шлак, штейн и т. п.).
[править]По повышению температуры кипения раствора
Температура кипения раствора 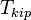 выше температуры кипения чистого растворителя
выше температуры кипения чистого растворителя 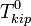 . Данные об изменении температуры кипения раствора могут быть использованы для расчета активности растворителя, в соответствии с уравнением:
. Данные об изменении температуры кипения раствора могут быть использованы для расчета активности растворителя, в соответствии с уравнением:
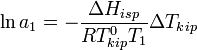 ,
,
где 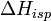 — теплота испарения растворителя, в интервале от температуры кипения чистого растворителя до температуры кипения раствора принимаемая постоянной. Индексом «1» обычно обозначается растворитель.
— теплота испарения растворителя, в интервале от температуры кипения чистого растворителя до температуры кипения раствора принимаемая постоянной. Индексом «1» обычно обозначается растворитель.
[править]По понижению температуры замерзания раствора
Температура замерзания раствора 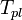 ниже температуры замерзания чистого растворителя
ниже температуры замерзания чистого растворителя 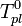 . Соответственно, активность растворителя можно рассчитать, используя зависимость:
. Соответственно, активность растворителя можно рассчитать, используя зависимость:
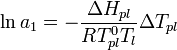 ,
,
где 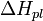 — теплота плавления растворителя.
— теплота плавления растворителя.
[править]По осмотическому давлению раствора
Величина осмотического давления раствора может быть использована для определения активности растворителя в соответствии с соотношением:
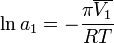
где 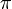 — осмотическое давление,
— осмотическое давление, 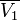 — парциальный молярный объём растворителя.
— парциальный молярный объём растворителя.
Коэффициент активности – коэффициент, связывающий реальную концертрацию электролита с его термодинамической активностью в уравнении a = γc (a – активность; с– концентрация; γ – коэффициент активности).
Необходимость введения активностей обусловлена отклонениями термодинамических уравнений от идеальности за счет электростатических сил и сил межмолекулярного взаимодействия. В этом случае при использовании концетраций результаты расчета не сответствуют экспериментальным данным.
Чтобы уравнения для идеальных растворов не меняли вид, в них используются активности. Для определения коэффициентов активности используются различного вида приближения, в основе которых лежит предельный закон Дебая-Хюккеля.
Сам по себе предельный закон Дебая-Хюккеля имеет ограниченное применение в области сильно разбавленных растворов, поэтому чаще пользуются различными усовершенствованными приближениями.
Одной из форм приближения является формула: [1, c.51]
lg γ± = — A |z+z–|I½ / (1 + I½) + CI,
где
γ± – среднеионной коэффициент активности;
A – константа, зависящая от диэлектрической проницаемости среды и от температуры [1, c.44];
I – ионная сила раствора;
C – эмпирическая константа, обычно принимаемая значение -0.1.
ДЕБАЯ - ХЮККЕЛЯ ТЕОРИЯ, статистич. теория разбавленных р-ров сильных электролитов, позволяющая рассчитать коэф. активностиионов. Основана на предположении о полной диссоциации электролита на ионы, к-рые распределены в р-рителе, рассматриваемом как непрерывная среда. Каждый ион действием своего электрич. заряда поляризует окружение и образует вокруг себя нек-рое преобладание ионов противоположного знака - т. наз. ионную атмосферу. В отсутствие внеш. электрич. поля ионная атмосфера имеет сферич. симметрию и ее заряд равен по величине и противоположен по знаку заряду создающего ее центр. иона. Потенциал суммарного электрич. поля, создаваемого центр. ионом и его ионной атмосферой в точке, расположенной на расстоянии r от центр.иона, м.б. рассчитан, если ионную атмосферу описывать непрерывным распределением плотности заряда около центр. иона. Для расчета используют ур-ние Пуассона (в системе СИ):
n2= /0,
где n2-оператор Лапласа, - диэлектрич. проницаемость р-рителя, 0 - электрич. постоянная (диэлектрич. проницаемость вакуума). Для каждого i-го сорта ионов описывается ф-цией распределения Больцмана; тогда в приближении, рассматривающем ионы как точечные заряды (первое приближение Дебая - Хюккеля теории), решение ур-ния Пуассона принимает вид:
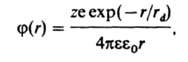
где z - зарядовое число центр. иона, rd - т. наз. дебаевский радиус экранирования (радиус ионной атмосферы). На расстояниях r > rdпотенциал становится пренебрежимо малым, т. е. ионная атмосфера экранирует электрич. поле центр. иона. Величина rd равна радиусу сферы, заряд к-рой равен заряду центр. иона и к-рая создает в месте нахождения центр. иона такой же потенциал, что иионная атмосфера; значение rd выражается ф-лой:
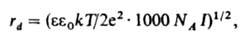
где k - постоянная Больцмана, Т - т-ра, NA - постоянная Авогадро, I - т. наз. ионная сила р-ра, зависящая от состава. Она определяется выражением:
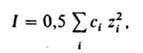
где сi - молярная концентрация i-го иона в моль/см3, zi - eгo зарядовое число; суммирование производится по всем типам ионов, присутствующих в р-ре. Дебая - Хюккеля теория дает возможность рассчитать средний ионный коэф. активности из выражения (предельный закон Дебая-Хюккеля):

где коэф. А выражается ф-лой:
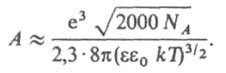
Для водных р-ров при 25°С А = 0,51. Согласно правилу Льюиса-Рендалла, коэф. активности данного типа ионов не зависит от типа др. присутствующих в р-ре ионов, а зависит только от ионной силы р-ра. Дебая - Хюккеля теорию широко используют для расчета коэф.активности ионов в разбавленных р-рах и концентрац. зависимости осмотич. коэффициентов. В первом приближении теория удовлетворительно описывает св-ва р-ров 1,1-валентных электролитов в области концентраций до 0,01 М, а для др. электролитов и неводных р-ров - в меньшем диапазоне концентраций. Введение поправок, учитывающих конечный размер ионов (второе приближение) и уменьшение вблизи ионов (третье приближение), позволяет применять Дебая - Хюккеля теорию в более широком диапазонеконцентраций; для водных р-ров 1,1-валентных электролитов - до 0,1 М. Ур-ние для расчета коэф. активности в третьем приближении Дебая - Хюккеля теории имеет вид:
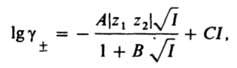
где В и С - эмпирич. постоянные. Ограниченность Дебая - Хюккеля теории обусловлена пренебрежением ассоциаций ионов, представлением о р-рителе как о непрерывной среде, характеризуемой только значением , т. е. неучетом мол. структуры р-рителя и его взаимод. с ионами. Дебая - Хюккеля теория является основой теории электропроводности разбавл. р-ров сильных электролитов, разработанной Л. Онсагером. Она позволяет объяснить увеличение электропроводности р-ра при повышении напряженности постоянного электрич. поля (эффект Вина) и в высокочастотном поле (эффект Дебая-Фалькенхагена). В этих условиях ионная атмосфера, тормозящая движение ионов, не успевает образоваться (см. Электропроводность электролитов). Теория создана П. Дебаем и Э. Хюккелем в 1923.
21 вопрос
Энергия активации, разность между значениями средней энергии частиц (молекул, радикалов, ионов и др.), вступающих в элементарный акт химической реакции, и средней энергии всех частиц, находящихся в реагирующей системе. Для различныххимических реакций Энергия активации изменяется в широких пределах - от нескольких до ~ 10 дж./ моль. Для одной и той жехимической реакции значение Энергия активации зависит от вида функций распределения молекул по энергиям их поступательного движения и внутренним степеням свободы (электронным, колебательным, вращательным). Как статистическую величину Энергия активации следует отличать от пороговой энергии, или энергетического барьера, - минимальной энергии, которой должна обладать одна пара сталкивающихся частиц для протекания данной элементарной реакции.
В рамках представлений теории абсолютных скоростей реакций Энергия активации - разность между значениями средней энергии активированных комплексов и средней энергии исходных молекул.
Представления об Энергия активации возникли в 70-80-х гг. 19 в. в результате работ Я. Вант-Гоффа и С. Аррениуса, посвященных изучению влияния температуры на скорость химической реакции. Константа скорости реакции k связана с Энергия активации (Е) уравнение м Аррениуса:
k = koe -E/RT
где R - газовая постоянная, Т - абсолютная температура в К, k o - постоянная, называемая предэкспоненциальным множителем константы скорости. Это уравнение, основанное на молекулярно-кинетической теории, позже было получено в статистической физике с учетом ряда упрощающих предположений, одно из которых - независимость Энергия активации от температуры. Для практики и для теоретических расчетов в сравнительно узких температурных интервалах это предположение справедливо.
Энергия активации можно найти по экспериментальным данным несколькими способами. Согласно одному из них, исследуют кинетику реакции при нескольких температурах (о методах см. в ст. Скорость химической реакции) и строят график в координатахIn k - 1 /T; тангенс угла наклона прямой на этом графике, в соответствии с уравнением Аррениуса, равен Е. Для одностадийных обратимых реакций (см. Обратимые и необратимые реакции) Э. а. реакции в одном из направлений (прямом или обратном) можно вычислить, если известна Энергия активации реакции в другом и температурная зависимость константы равновесия (из термодинамических данных). Для более точных расчетов следует учитывать зависимость Энергия активации от температуры.
Энергия активации сложных реакций представляет собой комбинацию Энергия активации элементарных стадий. Иногда, помимо истинной Энергия активации, определяемой по уравнению Аррениуса, используют понятие «кажущейся» Энергия активации Например, если константы скоростей гетерогенно-каталитических реакций определяют по изменению объемных концентраций исходных веществ и продуктов, то кажущаяся Энергия активации отличается от истинной на величину тепловых эффектов, сопровождающих процессы адсорбции и десорбции реагирующих веществ на поверхности катализатора. В неравновесных системах, например плазмохимических (см. Плазмохимия), определение Энергия активации является очень сложной задачей. В некоторых случаях, однако, возможно формальное применение уравнения Аррениуса
Уравне́ние Арре́ниуса устанавливает зависимость константы скорости химической реакции 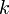 от температуры
от температуры 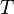 .
.
Согласно простой модели столкновений химическая реакция между двумя исходными веществами может происходить только в результате столкновения молекул этих веществ. Но не каждое столкновение ведёт к химической реакции. Необходимо преодолеть определённый энергетический барьер, чтобы молекулы начали друг с другом реагировать. То есть молекулы должны обладать некой минимальной энергией (энергия активации 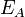 ), чтобы этот барьер преодолеть. Из распределения Больцмана для кинетической энергии молекул известно, что число молекул, обладающих энергией
), чтобы этот барьер преодолеть. Из распределения Больцмана для кинетической энергии молекул известно, что число молекул, обладающих энергией 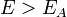 , пропорционально
, пропорционально 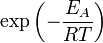 . В результате скорость химической реакции представляется уравнением, которое было получено шведским химиком Сванте Аррениусом из термодинамических соображений:
. В результате скорость химической реакции представляется уравнением, которое было получено шведским химиком Сванте Аррениусом из термодинамических соображений:
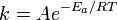
Здесь 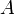 характеризует частоту столкновений реагирующих молекул,
характеризует частоту столкновений реагирующих молекул, 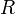 — универсальная газовая постоянная.
— универсальная газовая постоянная.
В рамках теории активных соударений зависит от температуры, но эта зависимость достаточно медленная:
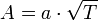
Оценки этого параметра показывают, что изменение температуры в диапазоне от 200 °C до 300 °C приводит к изменению частоты столкновений A на 10 %.
В рамках теории активированного комплекса получаются другие зависимости от температуры, но во всех случаях более слабые, чем экспонента.
Уравнение Аррениуса стало одним из основных уравнений химической кинетики, а энергия активации — важной количественной характеристикой реакционной способности веществ.
22 вопрос
Эбулиоскопия (от лат. ebulio — вскипаю) — метод исследования растворов, основанный на измерении повышения их температуры кипения по сравнению с чистым растворителем. Используется для определения молекулярной массы растворенного вещества, активности растворителя, степени диссоциации (или изотонического коэффициента).
Температура кипения жидкости — такая температура, при которой давление пара над жидкостью равно внешнему давлению. В то же время давление пара над раствором нелетучего вещества практически полностью определяется давлением пара растворителя и, в соответствии с законом Рауля, может быть выражено уравнением:
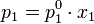
где x 1 — мольная доля растворителя.
Видно, что при повышении концентрации растворенного вещества давление пара над раствором будет снижаться, а следовательно, при неизменном внешнем давлении, будет расти температура кипения.
С учетом уравнения Клапейрона — Клаузиуса можно показать[1], что изменение температуры кипения раствора (Δ Tboil) может быть рассчитано по формуле:
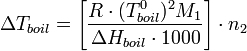
где Δ Hboil — энтальпия испарения;
M 1 — молярная масса растворителя;
n 2 — моляльная концентрация растворенного вещества.
Дробь в квадратных скобках в этом выражении зависит только от свойств растворителя — это так называемая эбулиоскопическая константа растворителя ε. Она равна повышению температуры кипения одномоляльного раствора.
Если известны изменение температуры кипения и концентрация раствора, можно определить молярную массу растворенного вещества:
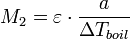
где a — число грамм растворенного вещества на 1000 г растворителя. Этот метод применим для разбавленных растворов нелетучих веществ и неэлектролитов.
Эбулиоскопический метод позволяет судить о состоянии вещества в растворах электролитов, так как для последних:
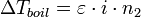 ;
;
где i — изотонический коэффициент.
С помощью эбулиоскопии можно определить и активность растворителя, в соответствии с формулой[2]:
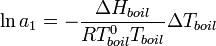
Криоскопия (от греч. κρύο — холод и греч. σκοπέω смотрю) — метод исследования растворов, в основе которого лежит измерение понижения температуры замерзанияраствора по сравнению с температурой замерзания чистого растворителя. Был предложен Ф. Раулем в 1882 году.
Давление пара над раствором нелетучего вещества практически полностью определяется давлением пара растворителя и может быть выражено уравнением (согласно закону Рауля):
где x 1 — мольная доля растворителя.
Видно, что оно ниже, чем давление пара над чистым растворителем, и снижается с ростом концентрации растворенного вещества.
В то же время при замерзании раствора давление пара над твердой фазой должно быть равно давлению пара над жидкостью. Если при замерзании раствора выделяется чистый растворитель, то давление пара над жидким раствором должно быть равно давлению пара над твердым чистым растворителем. Как было показано выше, давление пара над раствором ниже давления пара над чистым жидким растворителем, а следовательно, и соответствующее температуре замерзания равновесие для раствора будет устанавливаться при меньших температурах, чем для чистого растворителя. Это явление имеет важное значение в природе и технике.
Из приведенного выше выражения (закона Рауля), с учетом уравнения Клапейрона — Клаузиуса можно показать[1], что изменение температуры замерзания Δ Tcr для разбавленных растворов может быть рассчитано по формуле:
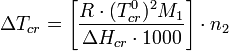
где Δ Hcr — энтальпия замерзания (кристаллизации);
M 1 — молярная масса растворителя;
n 2 — моляльная концентрация растворенного вещества.
Здесь выражение в квадратных скобках зависит только от природы растворителя — это так называемая криоскопическая постоянная растворителя k:
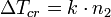
Для воды k = 1,84 K, для железа 110 K[1].
Измеряя Δ Tcr, можно определить молярную массу растворенного вещества, в соответствии с выражением:
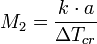
где a — число граммов растворенного вещества, приходящееся на 1000 граммов растворителя.
Криоскопия может быть использована для определения активности растворителя, в соответствии с соотношением[2]:
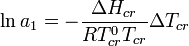
Кроме того, с помощью криоскопии можно определять степень диссоциации слабых электролитов, чистоту вещества, изучать комплексообразование в растворах и пр.
23 вопрос
Осмотическое давление (обозначается π) — избыточное гидростатическое давление на раствор, отделённый от чистого растворителя полупроницаемой мембраной, при котором прекращается диффузия растворителя через мембрану. Это давление стремится уравнять концентрации обоих растворов вследствие встречной диффузии молекул растворённого вещества и растворителя.
На примере воды появление осмотического давления обусловлено существованием полупроницаемых перегородок (мембран), которые пропускают отдельные молекулы воды, но препятствуют прохождению гидратированных ионов. Если подобная перегородка помещена между растворами разных концентраций, то из раствора меньшей концентрации в раствор большей концентрации будет переходить больше молекул воды, чем в обратном направлении. Возникает своеобразное явление перетекания воды, которое продолжается до тех пор, пока не произойдет выравнивание концентраций или пока этот процесс (если он протекает в ограниченном объеме) не будет уравновешен возникающим гидростатическим давлением. Такое давление называется осмотическим.
Раствор, имеющий более высокое осмотическое давление по сравнению с другим раствором, называется гипертоническим, имеющий более низкое — гипотоническим.
Осмотическое давление может быть весьма значительным. В дереве, например, под действием осмотического давления растительный сок (вода с растворёнными в ней минеральными веществами) поднимается по ксилеме от корней до самой верхушки. Одни только капиллярные явления не способны создать достаточную подъёмную силу — например, секвойям требуется доставлять раствор на высоту даже до 100 метров. При этом в дереве движение концентрированного раствора, каким является растительный сок, ничем не ограничено.
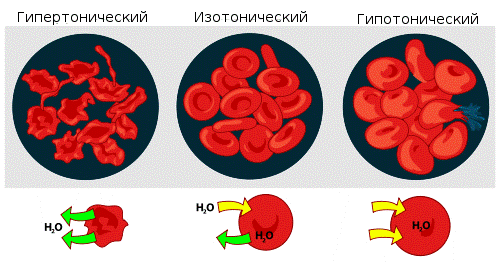

Взаимодействие эритроцитов с растворами в зависимости от их осмотического давления.
Если же подобный раствор находится в замкнутом пространстве, например, в клеткекрови, то осмотическое давление может привести к разрыву клеточной мембраны. Именно по этой причине лекарства, предназначенные для введения в кровь, растворяют в изотоническом растворе, содержащем столько хлорида натрия (поваренной соли), сколько нужно, чтобы уравновесить создаваемое клеточной жидкостью осмотическое давление. Если бы вводимые лекарственные препараты были изготовлены на воде или очень сильно разбавленном (гипотоническом по отношению к цитоплазме) растворе, осмотическое давление, заставляя воду проникать в клетки крови, приводило бы к их разрыву. Если же ввести в кровь слишком концентрированный раствор хлорида натрия (3-5-10 %, гипертонические растворы), то вода из клеток будет выходить наружу, и они сожмутся. В случае растительных клеток происходит отрыв протопласта от клеточной оболочки, что называется плазмолизом. Обратный же процесс, происходящий при помещении сжавшихся клеток в более разбавленный раствор, — соответственно,деплазмолизом.
Величина осмотического давления, создаваемая раствором, зависит от количества, а не от химической природы растворенных в нём веществ (или ионов, если молекулы вещества диссоциируют), следовательно, осмотическое давление является коллигативным свойством раствора. Чем больше концентрация вещества в растворе, тем больше создаваемое им осмотическое давление. Это правило, носящее название закона осмотического давления, выражается простой формулой, очень похожей на некий законидеального газа:
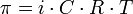 ,
,
где i — изотонический коэффициент раствора; C — молярная концентрация раствора, выраженная через комбинацию основных единиц СИ, то есть, в моль/м3, а не в привычных моль/л; R — универсальная газовая постоянная; T — термодинамическая температура раствора.
Это показывает также схожесть свойств частиц растворённого вещества в вязкой среде растворителя с частицами идеального газа в воздухе. Правомерность этой точки зрения подтверждают опыты Ж. Б. Перрена (1906): распределение частичек эмульсии смолы гуммигута в толще воды в общем подчинялось закону Больцмана.
Осмотическое давление, которое зависит от содержания в растворе белков, называется онкотическим (0,03 — 0,04 атм.). При длительном голодании, болезни почек концентрация белков в крови уменьшается, онкотическое давление в крови снижается и возникают онкотические отёки: вода переходит из сосудов в ткани, где πОНК больше. При гнойных процессах πОНК в очаге воспаления возрастает в 2-3 раза, так как увеличивается число частиц из-за разрушения белков. В организме осмотическое давление должно быть постоянным (≈ 7,7 атм.). Поэтому пациентам вводят изотонические растворы (растворы, осмотическое давление которых равно πПЛАЗМЫ ≈ 7,7 атм. (0,9 % NaCl — физиологический раствор, 5 % раствор глюкозы). Гипертонические растворы, у которых π больше, чем πПЛАЗМЫ, применяются в медицине для очистки ран от гноя (10 % NaCl), для удаления аллергических отёков (10 % CaCl2, 20 % глюкоза), в качестве слабительных лекарств (Na2SO4∙10H2O, MgSO4∙7H2O).
Закон осмотического давления можно использовать для расчёта молекулярной массы данного вещества (при известных дополнительных данных).
Диффузионное давление — давление, оказываемое диффундирующим веществом на воображаемую мембрану, помещенную в какую либо точку системы, где происходит диффузия. Чем ближе эта точка к источнику диффундирующего вещества — тем больше давление.
24 вопрос
Электролитическая диссоциация — процесс распада электролита на ионы при растворении его в полярном растворителе или при плавлении.
| Содержание [убрать] · 1 Диссоциация в растворах · 2 Диссоциация при плавлении · 3 Классическая теория электролитической диссоциации · 4 Сильные электролиты · 5 См. также · 6 Ссылки |
[править]Диссоциация в растворах
Диссоциация на ионы в растворах происходит вследствие взаимодействия растворённого вещества с растворителем; по данным спектроскопических методов, это взаимодействие носит в значительной мере химический характер. Наряду с сольватирующей способностью молекул растворителя определённую роль в электролитической диссоциации играет также макроскопическое свойство растворителя — его диэлектрическая проницаемость (Схема электролитической диссоциации).
[править]Диссоциация при плавлении
Под действием высоких температур ионы кристаллической решётки начинают совершать колебания, кинетическая энергия повышается, и наступит такой момент (при температуре плавления вещества), когда она превысит энергию взаимодействия ионов. Результатом этого является распад вещества на ионы.
[править]Классическая теория электролитической диссоциации
Классическая теория электролитической диссоциации была создана С. Аррениусом и В. Оствальдом в 1887 году. Аррениус придерживался физической теории растворов, не учитывал взаимодействие электролита с водой и считал, что в растворах находятся свободные ионы. Русские химики И. А. Каблуков и В. А. Кистяковский применили для объяснения электролитической диссоциации химическую теорию растворов Д. И. Менделеева и доказали, что при растворении электролита происходит его химическое взаимодействие с водой, в результате которого электролит диссоциирует на ионы.
Классическая теория электролитической диссоциации основана на предположении о неполной диссоциации растворённого вещества, характеризуемой степенью диссоциацииα, т. е. долей распавшихся молекул электролита. Динамическое равновесие между недиссоциированными молекулами и ионами описывается законом действующих масс. Например, электролитическая диссоциация бинарного электролита KA выражается уравнением типа:
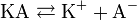
Константа диссоциации Kd определяется активностями катионов 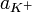 , анионов
, анионов  и недиссоциированных молекул
и недиссоциированных молекул 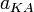 следующим образом:
следующим образом:
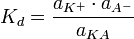
Значение Kd зависит от природы растворённого вещества и растворителя, а также от температуры и может быть определено несколькими экспериментальными методами. Степень диссоциации (α) может быть рассчитана при любой концентрации электролита с помощью соотношения:
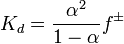 ,
,
где 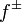 — средний коэффициент активности электролита.
— средний коэффициент активности электролита.
[править]Сильные электролиты
Классическая теория электролитической диссоциации применима лишь к разбавленным растворам слабых электролитов. Сильные электролиты в разбавленных растворахдиссоциированы практически полностью, поэтому представления о равновесии между ионами и недиссоциированными молекулами лишено смысла. Согласно представлениям, выдвинутым в 20—30-х гг. 20 в. В. К. Семенченко (СССР), Н. Бьеррумом (Дания), Р. М. Фуоссом (США) и др., в растворах сильных электролитов при средних и высоких концентрациях образуются ионные пары и более сложные агрегаты. Современные спектроскопические данные показывают, что ионная пара состоит из двух ионов противоположного знака, находящихся в контакте («контактная ионная пара») или разделённых одной или несколькими молекулами растворителя («разделённая ионная пара»). Ионные пары электрически нейтральны и не п

